


Abstract
Objectives: Characterization of the phenotypic, pathological, radiological, and genetic findings in 2 Saudi Arabian families with anoctaminopathies, and limb girdle muscular dystrophy type 2L (LGMD2L).
Methods: Over a 2-year period from December 2010 to January 2013, the clinical presentations were analyzed and all genes responsible for limb girdle muscular dystrophy (LGMD) were screened in families seen at King Faisal Specialist Hospital and Research Centre, a tertiary care hospital in Riyadh, Saudi Arabia. Out of 66 families with LGMD, we identified 2 families (3.1%) with anoctaminopathy, ANO5 muscular dystrophy.
Results: In the first case, a man presented with asymmetrical calves’ muscles weakness and atrophy, which was first noted at age 39. The creatinine kinase (CK) level was >20x normal, muscle biopsy showed necrotizing myopathic changes, and an MRI of the legs showed fatty-tissue replacement to muscle tissue with volume loss involving the gastrocnemius and soleus muscles in an asymmetrical fashion. Minimal disease progression was noted over 18 years of follow up. Exercise induced recurrent rhabdomyolysis was noted over the last 2 years. A novel ANO5 gene mutation (Arg58Trp) was found. In the second family, a male presented at the age of 41 with asymptomatic hyperCkemia and intermittent dyspnea. Over 10 years follow up, he became disabled with muscle cramps, rhabdomyolysis, myoglobinurea, and difficulty ambulating. Muscle biopsy showed necrotizing myopathy and perivascular and interstitial amyloid deposit in skeletal muscle. A homozygous deletion of 11.9 Kb encompassing exon 13 to exon 17 was found in the ANO5 gene. Full cardiac investigations were normal in both patients.
Conclusion: The prevalence of LGMD2L is approximately 3.1% in a Saudi Arabian native LGMD cohort. Slowly progressive, late onset, and asymmetrical weakness was the salient features in these 2 families. The genetic findings were novel and will add to the spectrum of ANO5 known mutations.
Anoctamin is a protein involved in the calcium-activated chloride channel and named as such because it contains 8 transmembrane domains that are anions (ano “anion” and octa=8). The exact function of anoctamin 5 (ANO5) proteins are still not exactly known. It is possibly involved in cell membrane repair.1 An autosomal recessive mutation in ANO5 causes limb girdle muscular dystrophy type 2L (LGMD2L). This condition is characterized by proximal weakness affecting the pelvic girdle and leg muscles, or with only distal weakness known as non-dysferlin Miyoshi muscular weakness type 3 (MMD3).2-4 Anoctamin related muscle disorder was first described in 14 French Canadian patients in 2007.2 Subsequently, only a few cases were described from different ethnic groups.3-15 Cases of isolated hyperCKemia and pseudo metabolic myopathy were also reported to be caused by ANO5 gene mutations (MIN# 608662). In this communication, we describe the clinical and genetic findings in 2 native Arab patients who presented with a long history of exercise intolerance and high CK, and were found to have ANO5 related muscular dystrophy.
Methods
This study was ethically approved and conducted at King Faisal Specialist Hospital and Research Centre (KFSH&RC), a tertiary care hospital in Riyadh, Saudi Arabia, which receives referrals from the whole country of approximately 25 million inhabitants. All cases diagnosed with limb girdle muscular dystrophy (LGMD) from the neuromuscular database over the period of December 2010 to January 2013 were studied. We were able to identify and enroll 66 families; 142 patients underwent neurological examination, skeletal muscle imaging, muscle biopsy, and cardiac evaluation. Two unrelated families included in this study were the subjects of this paper.
Homozygosity mapping
The DNA was extracted from peripheral blood samples using standard procedures (Flexi Gene DNA Handbook, Qiagen, Hilden, Germany). All participants were genotyped using the Affymetrix Cyto v.2 array (Affymetrix, Santa Clara, CA, USA) following the manufacturer’s protocol (http://www.affymetrix.com/support/technical/manuals.affx). Genotype results were analyzed using Chromosome Analysis Suite (ChAS) software (Affymetrix, Santa Clara, CA, USA). In these families, homozygosity testing of the affected individual was used to select candidate genes from 22 known LGMD loci.
Multiplex polymerase chain reaction
Exons 13-17 of ANO5 were amplified in conventional multiplex PCRs together with MYO5B exon 28, which was used as a reference for patient 2, and a normal control. Subsequently, the PCR products were subjected to electrophoresis in 2% agarose gel dissolved in Tris-EDTA buffer and DNA 1kb ladder marker was used as a reference for DNA sizing.
The DNA sequencing
The coding regions of candidate genes were amplified and sequenced using a BigDye Terminator kit and run on an ABI 3730xl automated sequencer (Applied Biosystems, Foster City, CA, USA). SeqScape v.2.6 software (Applied Biosystems, Foster City, CA, USA) was used to align sequence data.
Results
Patient 1
A Saudi man was asymptomatic until the age of 38 when he noticed that his right leg was bigger than the left without muscle cramps or weakness. A few months later, he had an episode of shortness of breath and epigastric pain. Cardiac investigations were normal but creatine phosphokinase (CK) was 9,000 IU (normal ≤ 185 IU); the chest pain disappeared within an hour. His disease progressed slowly over 18 years of follow-up and at age 59, he was still ambulatory with no walking aid. He mentioned he has limited walking tolerance with frequent rhabdomyolysis and myoglobinuria after moderate physical activity. His examination showed normal ocular, bulbar, and neck muscles, moderate atrophy of the leg muscles affecting the gastrocnemii, and hamstring muscles. The atrophy was asymmetrical affecting left more than the right (Figure 1A). The knees were markedly hyperextended (Figure 1B). The deltoids and triceps were minimally weak as well as the forearm extensors. The hamstring and calf muscles were markedly weaker than the anterior compartment of the lower limb, he could not stand on heels or toes; however, he could stand from sitting position with no hands support. The muscle biopsy showed severe necrotic myofibers, few inflammatory cells and normal Dysferlin staining and other sarcolemmal membrane staining. An MRI carried out twice at the age of 39 and 45 showed myoedema initially in the gastrocnemii and hamstring muscles, and subsequently severe fatty and fibrous tissue replacement in these muscles.
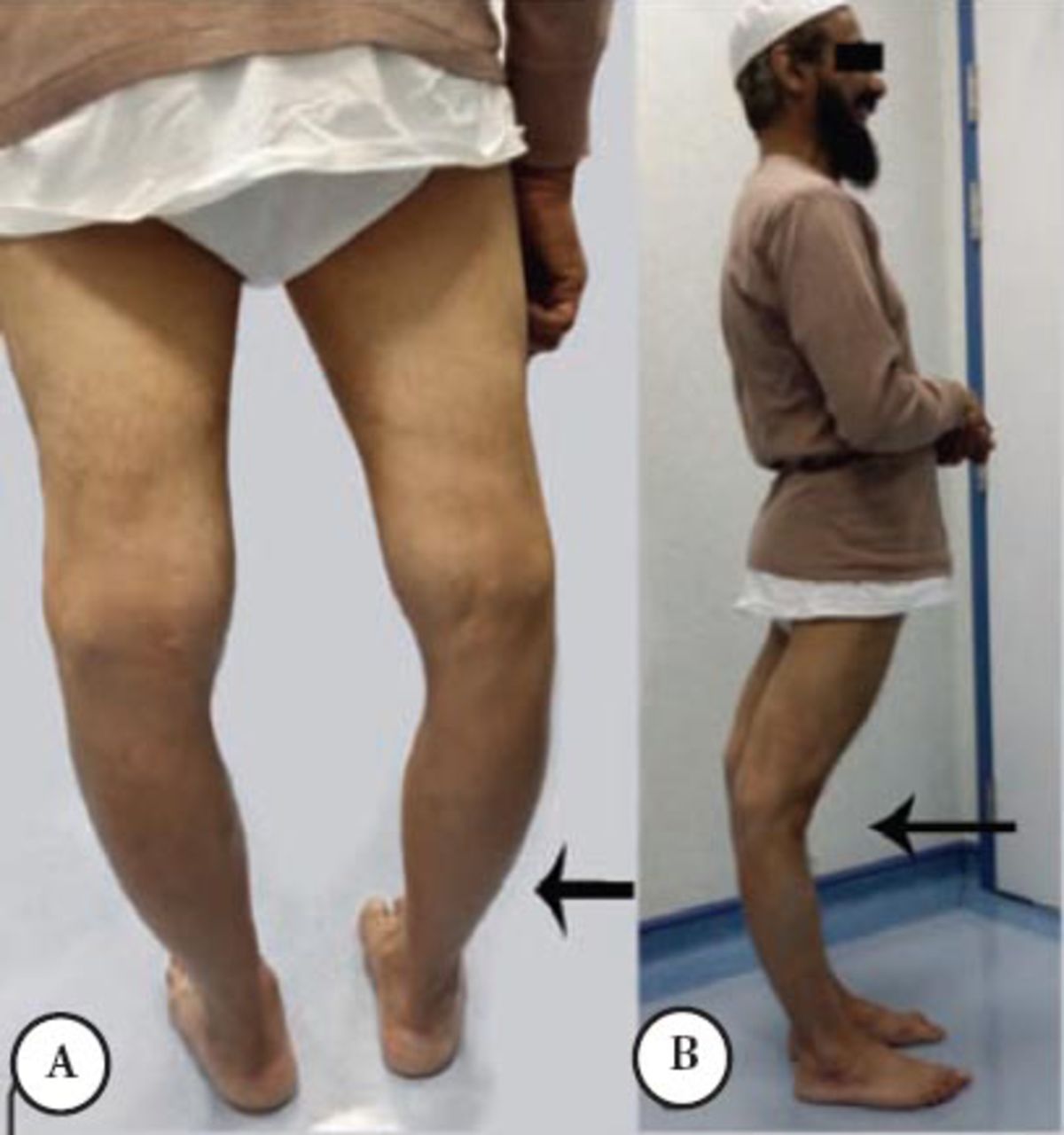
First patient A) showing prominent quadriceps and calf muscles asymmetrical atrophy (arrow head) and, B) clear knee hyperextension.
Patient 2
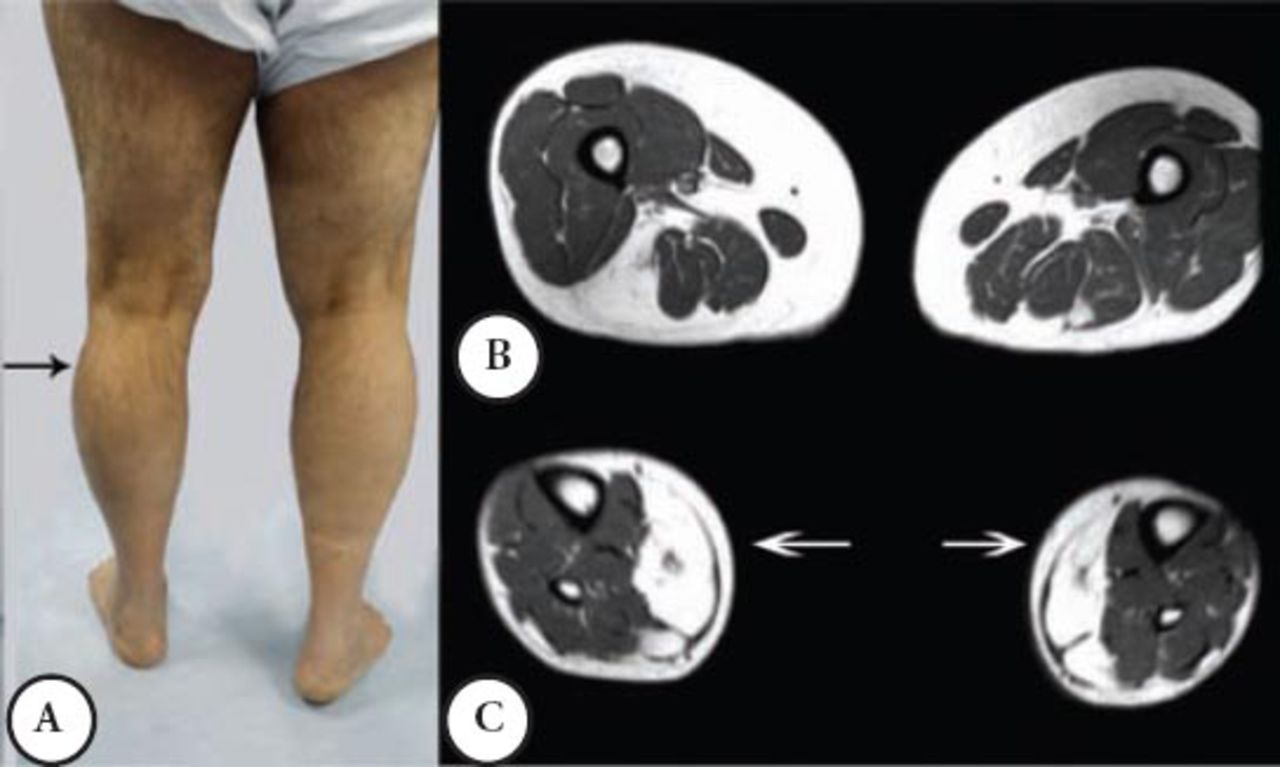
A male patient was well until the age of 38 when suddenly he had shortness of breath and was admitted to a coronary care unit. He was found to have a CK of 3,200 (normal ≤ 185 IU), coronary angiography and other cardiac investigations were normal; the shortness of breath was improved spontaneously. A similar incident occurred a few times over 3 years. He then developed intermittent exercise related rhabdomyolysis and myoglobinuria associated with muscle cramps. Since then, he had difficulty walking and his other activities like standing from a chair or climbing the stairs became more difficult and disabling. He required a cane for ambulation, and had to quit his job as a Banker at the age 48. He was seen by the Rheumatology Service, and erroneously he was diagnosed to have an inflammatory myopathy and received steroid therapy and intravenous immunoglobin for 2 years with no improvement. Currently, he is homebound and needs 2 people to help him stand; he can only walk for a few meters because of muscle cramps and pain. His parents are first-degree cousins, he and his wife are cousins, and they have 3 boys and one girl; none have a similar complaint. Mild global weakness of the proximal muscles affected the left more than the right. Minimum weakness was noted in the hands and forearm muscles. There was a bilateral and asymmetrical hypertrophy of the calf muscles mainly in the outer compartment compared with the inner compartment. He could stand on his heels but not on his toes, because of pain, he could not squat or rise from a low chair. The distal aspects of his leg were noticeably small and thin (Figure 2A), the knees were slightly hyper extendable. His CK was persistently high between 1,600 IU to 8,800 IU, muscle biopsy showed myopathic changes with evidence of denervation atrophy and reinnervation as well as some amyloid deposition in the perivascular and interstitial tissue. Immunohistochemistry showed normal sarcolemmal immunoreactivity with normal Dysferlin staining. Serum and urine protein electrophoresis and immunofixation, serum kappa, and lambda free light chains were normal. Staining for amyloid from abdominal fat tissue was negative. An MRI of the thigh showed moderate atrophy of the lateral and posterior muscle compartments with fatty replacement. The sartorius and gracilis muscles were selectively spared, in the leg MRI; fatty replacement affected the lateral compartment (Figures 2B & C).
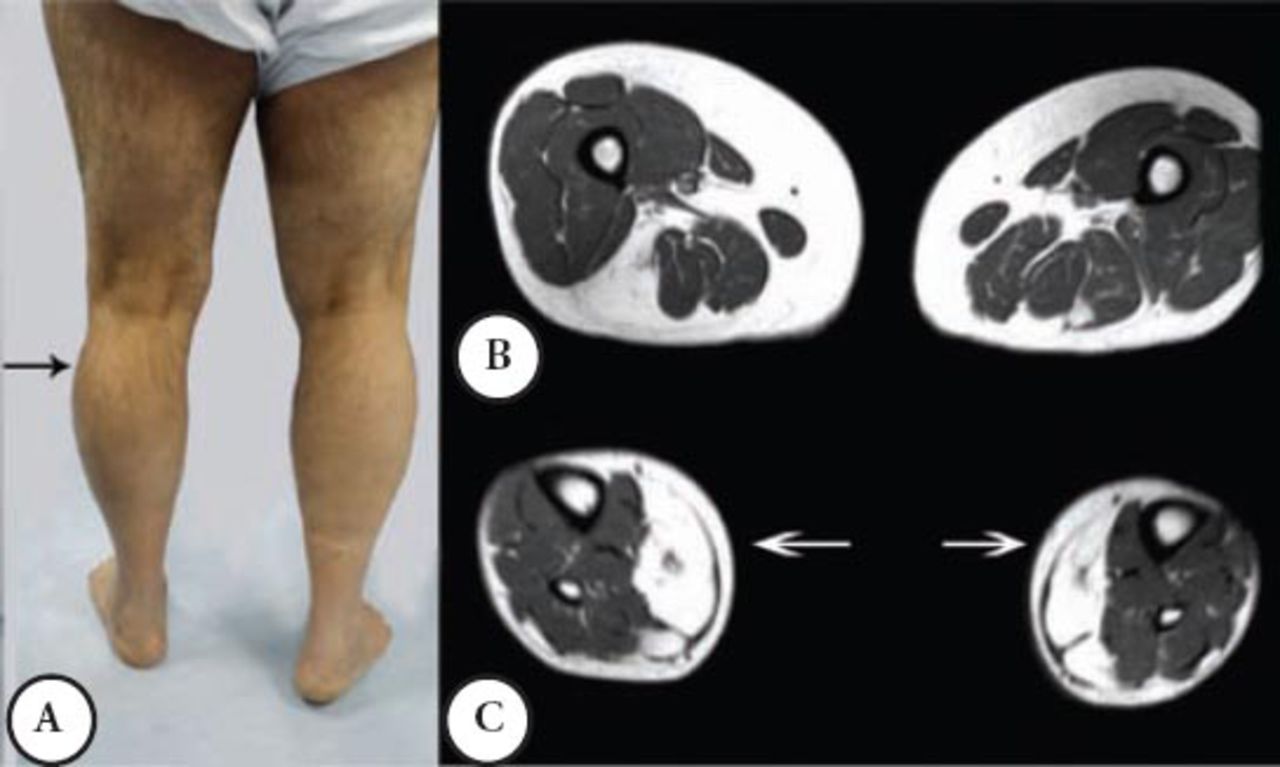
Second patient A) noted asymmetrical atrophy in the leg medial quadriceps and calf muscles. B) An MRI at the mid thigh region noted posterior and lateral muscles group atrophy and fatty infiltration. C) An MRI at mid leg region noted the mild asymmetrical fatty infiltration.
Genetic findings
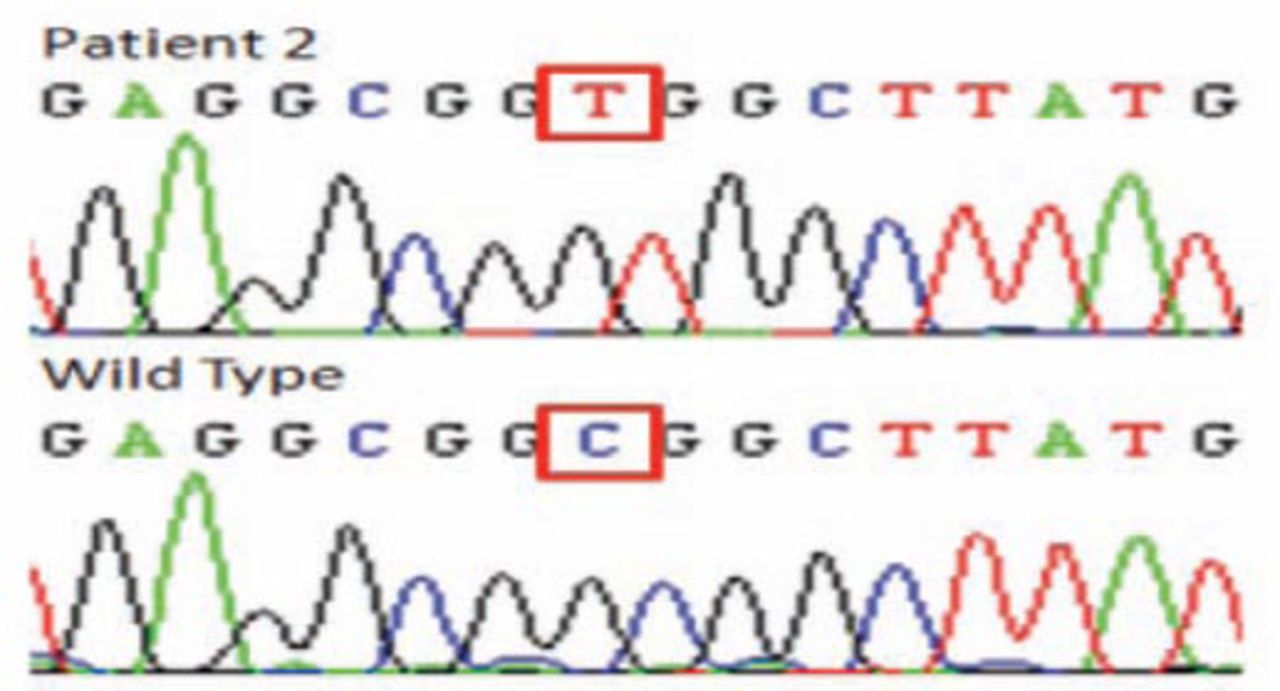
In patient one and 2, homozygosity mapping revealed 3 (DYSF, SGCA, ANO5) and 2 (FKRP, ANO5) candidate genes. In patient one, the sequencing results identified a novel missense mutation in exon 4 (c. 169C>T, p.R58W) (Figure 3) altering a topological domain of ANO5 predicted to be in the cytoplasmic region of this transmembrane protein. This missense mutation was absent in 200 unrelated Saudi individuals. The change was also predicted to be pathogenic by Polyphen2 (http://genetics.bwh.harvar.edu/pph2/) and SIFT (http://sift.bii.a-star.edu.sg/) with scores of 1.00 and 0.02.
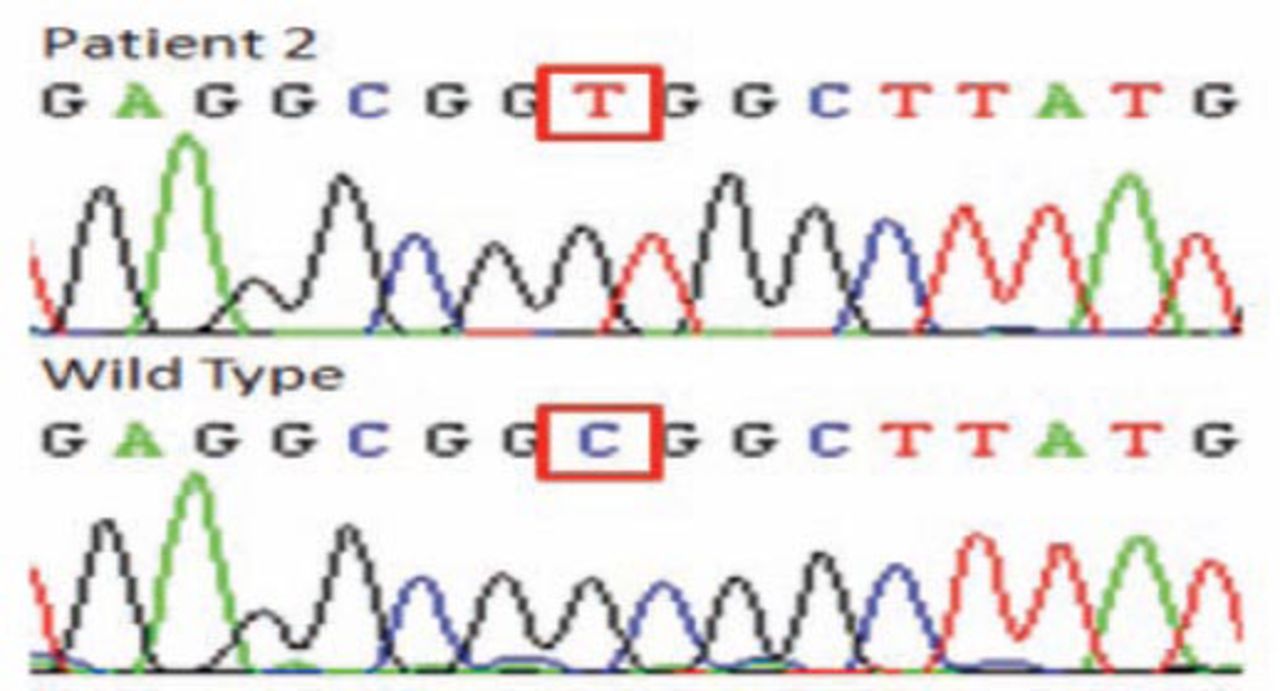
The DNA elecrophoregram with missense c.169C >T mutation, (p.R58W) of patient one.
In patient 2, a homozygous deletion encompassing exon 13 to 17 was detected using ChAS (Figure 4A). The absence of ANO5 exons 13 to 17 was confirmed by multiplex PCR (Figure 4B), and in the absence of nonsense mediated decay, is predicted to delete amino acid 394-633 encompassing several topological (both cytoplasmic and extracellular) and transmembrane domains of ANO5. A similar finding was reported previously.15

Genetic findings in patient 2 showing A) Copy number analysis using Chromosome Analysis Suite in patient 2 deletion containing exons: 13-17 of ANO5. B) Multiplex PCR including ANO5 (exons: 13,14,15,16,17) and ATL1 (Ex.6) as a internal control in patient one and normal control. ANO5 - anoctamin 5, PCR - polymerase chain reaction
Discussion
The 2 described patients had a peculiar presentation of hyperCKemia discovered incidentally after a transient period of dyspnea, followed by rhabdomyolysis, myalgia, and muscle cramps. These 2 patients were identified out of a cohort of 66 families with LGMD representing 3.1% of our LGMD cases. The prevalence of ANO5 related muscle disease is variable among different ethnic groups, ranging from 2×105 in Finland,9 and 1×105 in Denmark,5 or as low as 0.26×105 in the north of England.6 Among cohorts of LGMD cases, ANO5 was seen in 11% in the Netherlands,7 and 3.8% in Italy.8
The ANO5 related muscle disease ranges from asymptomatic hyperCKemia to a slowly progressive adult onset muscle weakness, which can be either proximal or distal.6,9,10 Another less common, but well-described presentation is an asymmetrical posterior leg compartment weakness and atrophy, as seen in our first patient, initially described as non-Dysferlin Miyoshi phenotype with distal weakness.9,10 In our patients, little disease progression was noted over 20 years of follow-up; both patients were still partially independent in their ambulation. Such mild disease progression was similarly reported in the literature.9 Rhabdomyolysis is a common feature of this disease and may occur at different stages.9 In a recent large reported cohort of 42 patients with anoctaminopathy from 42 European Centers,10 a predominantly proximal involvement was observed in 57% of patients, distal involvement in 10%, proximal plus distal involvement in 8%, and none, or very mild clinical symptoms (myalgia and/or raised CK) in 25%. Most of their patients were male. An exon 5 founder mutation (c.191dupA) has been confirmed to be the most common mutation.6
Both our patients had the similar initial phenomena of transient difficulty breathing and shortness of breath, but this complaint disappeared completely during their follow-ups. This is a unique observation as respiratory difficulties were not previously noted. However, in a Danish group of patients, subjective dysphagia was reported in 8 out of 20 patients.5
The number of recessive mutations in LGMD2L is growing rapidly,10,12 as described in German, Italian, Finnish, and Danish families; but rarely described in native Arabs.14 The mutation noted in patient one (Arg58Trp) was novel (Figure 3), and the common homozygous deletion encompassing exon 13 to exon 17 found in patient 2 (Figure 4) was reported previously.14
In LGMD2L, an MRI of the muscle shows fatty infiltration and dystrophic changes in the lateral and posterior compartment of the lower extremity, but notably sparing the sartorius and gracilis muscles.12 Myoedema and widely distributed hyperintense signal in the early stage of the diseases were seen on short tau inversion recovery (STIR) weighted images, with asymmetrical muscle improvement seen in one of our patients as reported previously.12 The muscle biopsies in our patients showed non-specific degenerative myopathy with scattered myofiber necrosis, and a few inflammatory cells, which led to an initial diagnosis of inflammatory myopathy in our 2 cases, a wrongly inflicted diagnosis commonly seen in this muscle disorder.13 Interestingly, our second patient had amyloid deposition in the muscle fibers and blood vessel in his muscle biopsy with no evidence of systemic amyloidosis with extensive investigation. This observation has been reported before, and described in muscle biopsies of the 2 types LGMD2L and LGM2B.15
In conclusion, we describe the clinical and genetic findings of 2 cases of ANO5 related muscle disease among Saudi Arabian patients, and add the novel genetic findings to the known previously reported mutation list.
REFERENCES
* References should be primary source and numbered in the order in which they appear in the text. At the end of the article the full list of references should follow the Vancouver style.
* Unpublished data and personal communications should be cited only in the text, not as a formal reference.
* The author is responsible for the accuracy and completeness of references and for their correct textual citation.
* When a citation is referred to in the text by name, the accompanying reference must be from the original source.
* Upon acceptance of a paper all authors must be able to provide the full paper for each reference cited upon request at any time up to publication.
* Only 1-2 up to date references should be used for each particular point in the text.
Sample references are available from:
Footnotes
Disclosure
Funding was received from King Abdulaziz City for Science and Technology Research No. 08-MED498-20.
- Received August 27, 2014.
- Accepted March 16, 2015.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.