


Abstract
Homozygous or compound heterozygous pathogenic variants of the RBCK1 gene can result in a systemic disorder characterized by the accumulation of complex carbohydrate molecules, namely polyglucosan bodies in the muscular tissues. The role of this gene in the pathophysiology of the disorder at the molecular level remains unclear. Being a very rare disorder, the medical knowledge is based on just a few reported cases. Here we report a 7-year-old girl who presented with exercise intolerance and hepatosplenomegaly. Her liver profile was constantly raised. The genetic investigation has revealed a variant of the RBCK1 gene of unknown significance, which has later been confirmed as pathogenic via a variety of clinical, genetic, and histopathological approaches. More importantly, it is evident that the availability of sophisticated genetic testing, such as whole-exome sequencing, has significantly improved the knowledge of and diagnosis of many rare metabolic disorders.
The polyglucosan bodies (PGs) are complex unspecific polysaccharide molecules that are resistant to alpha-amylase enzyme digestion. The PGs result from a defective metabolism of glycogen in some glycogen storage disorders, which subsequently causes the pathological accumulation of these bodies.1 Furthermore, PG consists of an excess of long peripheral chains with fewer points for branching as compared to the spherical structure of glycogen. The PG precipitate and build up in insoluble deposits that can be easily recognized via histopathology and electronic microscopic analysis.2 Individuals affected with RBCK1 mutations have been reported to have variable manifestations in regard to the predominant clinical phenotype. Some have muscular involvement as the hallmark symptoms of the disease, whereas others may present primarily with autoinflammation or immunodeficiency. The reason for this wide clinical spectrum is still unknown. However, it is speculated that there is a genotype-phenotype association, with a mutation in the N-terminal site of RBCK1 predisposing the patients to immunological disorder and mutations in the middle or C-terminal regions causing the myopathic subset.3 In this report, our patient presented with muscular myopathy and hepatomegaly without immunological or cardiomyopathy. She has a homozygous variant c.913C>T in exon 7 of the RBCK1 gene. Family segregation and muscle biopsy data supported the notion that this variant is pathogenic. This variant has not previously been reported although it is in dbSNP database with an rs1288748870.
Case Report
Patient information
Our patient is a 7-year-old girl born to relative parents. She is the 3rd in order. The patient presented first to a pediatric clinic at 3.5 years old with a history of abdominal distension and exercise intolerance. There was no similar history in the family. There was no history of jaundice, bleeding tendency, hypoglycemia, or fever. She has a history of acute tonsilitis and mouth breathing.
Clinical findings
During her first visit to the metabolic/genetics clinic at 3.5 years old, upon examination, she had normal growth and development. Her weight was 14.5 (25th percentile), height 94.4 cm (10th–25th percentile), and head circumference 50.5 cm (10th–25th percentile). She had enlarged tonsils and was a mouth breather. Air entry was normal with no wheezing. She had normal first and second heart sounds, no added sounds, and no murmurs. Her abdomen was soft and lax, and her liver was enlarged (to 6–7 cm below the costal margin). Her spleen was with the same measurement, with no shifting dullness. The patient was cooperative and oriented, with intact cranial nerves examination. She has hypotonia. There was mild weakness of the proximal muscles (power 4/5 bilaterally). Deep tendon reflexes were diminished and no clonus. The labs showed the followings as illustrated in Table 1.
- Laboratory results.
By ultrasound (US), the liver measured approximately 9.8 cm and was diffuse with increased echogenicity and small rounded echogenic lesions of approximately 0.6×0.7 cm involving the right hepatic lobe. The portal and hepatic veins were patent with the normal flow direction. The portal vein’s maximum velocity was 27.7 cm/s. The gallbladder appeared adequately distended and echo-free. We detected no biliary ductal dilatation. The spleen measured about 10.9 cm at the bipolar span. No gross focal splenic lesions could be seen.
Therapeutic intervention
The patient was kept for observation. We detected no hypoglycemia or deterioration of her hepatic or coagulation profile. The enzymes were trending down without any kind of intervention.
Diagnostic assessment
Histopathology of the right thigh muscle: Polyglucosan body disease and slight reinnervated skeletal muscle. In PAS-reacted sections, some fibers display multiple punctate accumulations of PAS-positive material that is partially digested by PAS diastase, consistent with polyglucosan bodies.
Echocardiogram (ECHO)
The patient had normal heart function, with only trivial mitral valve regurgitation. The EEG was normal, and no cardiomyopathy was detected.
Whole exome sequencing
RBCK1 (NM_031229.3):c.913T>C p.(Cys305Arg) homozygous
The variant classification based on ACMG recommendations: Class 3 which is variant of unknown significance VUS. This means the variants for which not all details are available. The reasons for that include unsequenced deletion or duplication breakpoints, variants reported on the protein level only or other variants likely affect the RNA splicing sites without RNA analysis. The genomic variant is NC_000020.10:g.401671T>C. The predictive tools support the pathogenic candidacy of our variant as follow:
-Polyphen: probably damaging
-SIFT: deleterious
-Align-GVGD: C65 which indicates the variant interferes with the function
-MutationTaster: Disease causing
-Conservation: amino acids not high vs high.
The pathogenicity verdict analysis via Varsome has revealed the followings: PP3 and PM2 indicating the pathogencity of the variant; The family segregation analysis as shown in Figure 1 reveled the homozygousity only detected in the patient whereas the other family members are asymptomatic heterozygous; The RBCK1 variant detected by whole exome sequencing and confirmed by NGS method.
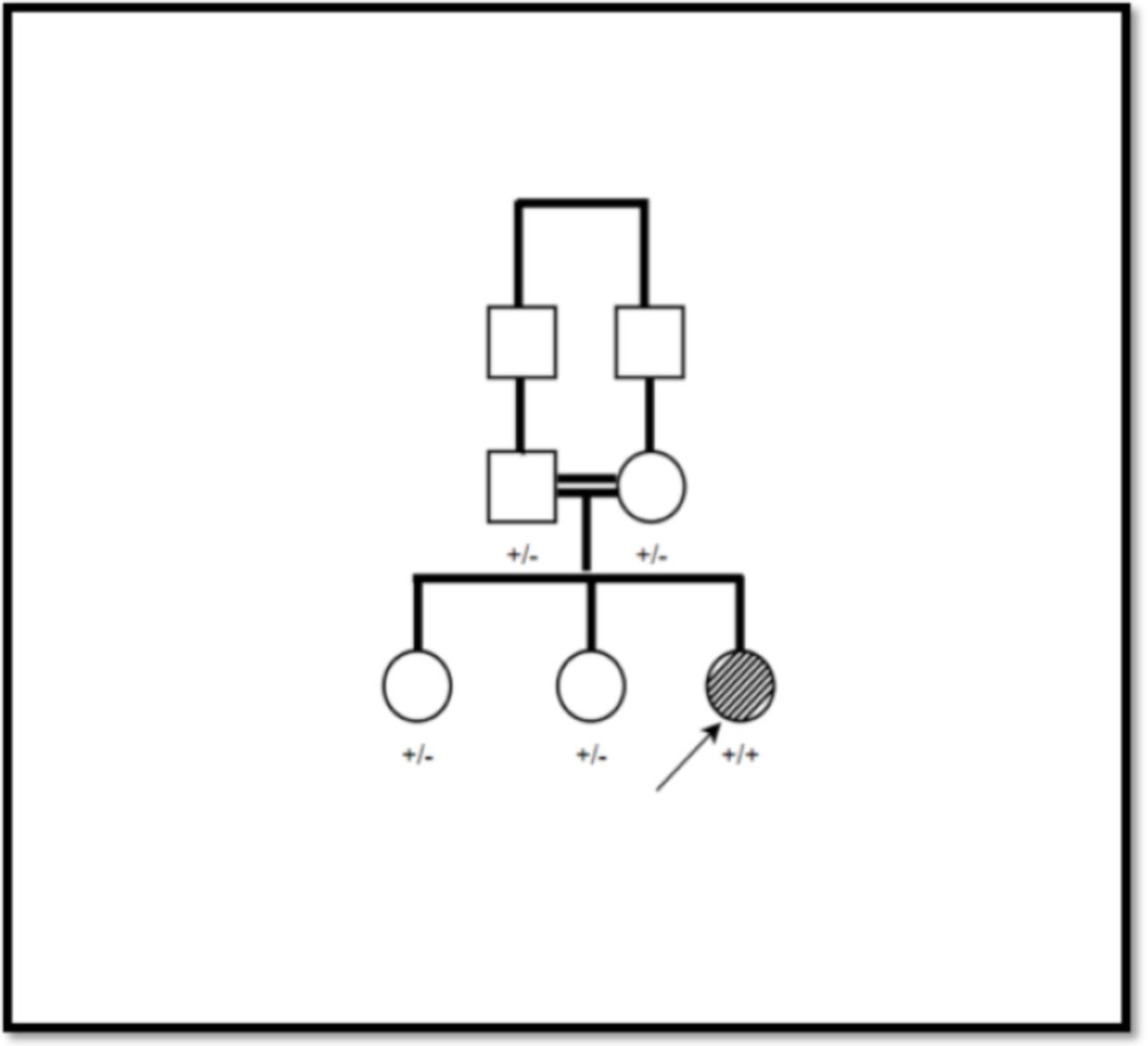
- family pedigree and segregation analysis, + indicates the mutant allele, - indicates the wild type allele
Follow up and outcome
The patient was regularly followed up in the metabolic and gastroenterology clinic. At 7 years old, she has no developmental delay or growth retardation. The liver enzymes remained elevated as follows: alanine transaminase (51 H, 5–33 U/L), aspartate transaminase (48 H, 5–32 U/L), normal coagulation and albumin level. As reported by her family, she has exercise intolerance if she makes any exertional effort, such as walking for long distances, but this generally does not halt her overall motor capabilities (Table 2).
- Timeline.
Discussion
In this report, we have described a 7- year-old girl with hepatomegaly and mild proximal myopathy. The genetic investigation has yielded a RBCK1 gene variant that is consistent with the phenotype, segregation analysis, and histopathological features of the polyglucosan myopathy disorder.
Based on previously reported cases in the medical literature, there is an apparently remarkable association between the RBCK1 variant exon site and the development of either immunodeficiency or myopathic phenotype. Biosson et al4 reported patients with disease-causing variants in the N-terminal part of RBCK1 with severe early-onset immunodeficiency that resulted in death at infancy. However, the patients reported by Nilsson et al. and Wang et al. have mutations either in the middle or C-terminal site of the RBCK1 gene and developed neuromuscular phenotype. It was later suggested that the nature and loci of the underlying disease-causing variant might determine the predominant clinical picture, with N-terminal disease-causing variants mainly causing immunological dysfunction, whereas variants in the middle- or C-terminal regions were presumed to predominantly cause cardiomyopathy and neuromuscular symptoms (Table 3).5
- RBCK1 genotype and phenotype association. Permission taken from the author as well as the journal
Additionally, the RBCK1 gene encodes heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1) and is related to HOIL-1L interacting protein (HOIP), which forms the linear ubiquitination chain assembly complex (LUBAC), a component of the NF-κB cascade involved in IKK complex activation. NF-κB has a significant impact on the regulation of the immune system. Therefore, disease-causing mutations in RBCK1 can lead to autoinflammation as well as immunodeficiency.6
Considering all cases reported to date, it appears that pathogenic RBCK1 variants invariably cause myopathy, but not necessarily immunological symptoms. So far, severe immunological phenotypes have only been reported for protein damaging N-terminal mutations.
A detailed review of the literature alongside newly reported cases shows that frameshift mutations beyond the N-terminus of RBCK1 may lead to a combined phenotype, including both myopathy and immunological dysfunction in single families. Our patient has a variant in exon 7 in the middle of the RBCK1 gene and exhibiting visceral involvement in the form of hepatomegaly without cardiomyopathy. Furthermore, there is no evidence of immunodeficiency, which supports this assumption of loci associated with the hallmark presentation. The c.913 C>T variant has not been reported in the literature; therefore, its novelty is considered in this paper.
Based on the clinical information, specific attention was paid to the genes in the lysosomal storage disease panel included in the WES analysis, these are: ARSA,FUCA1,GALC,GBA,GLB1,GNPTAB,GUSB,HEXA,HEXB,MAN2B1,MANBA,NAGA,SMPD1. No relevant variant in these genes was detected. For these genes, an overall coverage of 97.50% was achieved (>20x), with 683 missing base pairs (coding region including +/- 2bp). The whole exome sequencing data focusing on variants affecting protein function were analyzed (nonsense, frameshift, conserved splice site and missense with high pathogenicity predictions) in genes with supporting evidence from zygosity or segregation and additional evidence for a functional importance of the gene in the described phenotype based on available expression, experimental data or animal models.
The clinical usefulness of next-generation sequencing and whole exome techniques in complex muscular-neurogenetic diseases is of paramount value, and clinical correlations with genotypes may help in determining the mode of workup and intervention.
Acknowledgment
The authors gratefully acknowledge the histopathology lab for detecting the peculiar finding of polyglucosan bodies in the muscle tissues. To the authors and journals that allowed the re-use of published data.
- Received December 9, 2021.
- Accepted August 24, 2021.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}