


Abstract
Both malformations of the central nervous system and neurometabolic disorders are common, mainly in highly consanguineous populations. Both metabolic pathways and developmental pathways are closely related and interact with each other. Neurometabolic disorders can lead to disturbances in brain development through multiple mechanisms that include deficits in energy metabolism, critical nutrient deficiency, accumulation of neurotoxic substrates, abnormality in cell membrane constituents, and interference in cell-to-cell signaling pathways. The anomalies observed include absent or hypoplastic corpus callosum, midline brain defects, and malformations of the cortex, the cerebellum and the brain stem. Early diagnosis of an underlying inherited neurometabolic disorders is critical for the institution of treatment, which may positively influence prognosis, and allow for proper genetic counseling. In this review, we discuss those disorders in which the structural brain malformation is a dominant feature, and propose a practical approach that will permit a physician to investigate, and treat these disorders.
Both malformations of the central nervous system (CNS) and inherited neurometabolic disorders are common, mainly in highly consanguineous populations.1,2 Many neurometabolic disorders are well known to be associated with structural brain defects. peroxisomal disorders, mucopolysaccharidoses, and Smith-Lemli-Opitz syndrome are among the earliest examples of metabolic disorders with significant dysmorphology profiles.3 However, large studies are lacking, and the literature contains only a few small series of patients and many individual case reports.4-12 One possible explanation is that, in patients with developmental disorders, intellectual disability, or epilepsy, malformation of the CNS system serves as satisfactory explanation for the clinical manifestations, and these imaging findings might discourage some clinicians from further investigating for possible underlying diseases.
Advances in neuroimaging, particularly MRI/MR spectroscopy, now enable the diagnosis of malformations of the CNS and may offer important clues for any underlying neurometabolic disorders.13,15 Furthermore, advances in genetics, and molecular biology have improved our understanding of the control of CNS development and maturation. Metabolic and developmental pathways are closely related and interact with each other. Though an acquired condition, folic acid deficiency and malformation of the neural tube highlight the close relation between metabolism and structural defects. Folate is known to play a fundamental role in embryonic development. Experimental studies demonstrate that inactivation of genes in the folate pathway results in malformations of the neural tube, and craniofacial structures. The use of folic acid containing supplements in the periconceptional period decreased the occurrence of neural tube defects, and possibly other birth defects in the offspring of women, by 40-60%.16
Careful assessment of patients with structural brain defects and the recognition of possible underlying inherited metabolic disease are important to providing a possible treatment, an accurate prognosis and genetic counseling. The most recent literature regarding mechanisms of brain development and malformations in the context of neurometabolic disorders, and future research are discussed.
Normal brain development
Congenital anomalies of the CNS are extremely complex and are best studied by correlation with embryological development. Modern neuroembryology integrates descriptive morphogenesis about the parts of the CNS with insights into molecular genetic programming and data enabled by cell-specific tissue. Although an extensive review about this topic is not presented in this article, the basic events in normal brain development including the four stages, are summarized in Table 1.
Normal brain development.
Mechanisms of malformation of the CNS in inherited metabolic diseases
Most malformations of the CNS are best understood in the context of aberrations of normal developmental processes that result in abnormal structure and function. The cause of structural brain defects can be infectious, genetic/metabolic, environmental, or multifactorial.17 Early malformations are usually disorders of genetic expression along gradients of the three axes of the neural tube, defective segmentation, or mixed lineages of individual cells. Later, disorders mainly involve cellular migrations, axonal path finding, synaptogenesis, and myelination. There is a close link between the in utero metabolic environment, including adaptation of energy, carbohydrate, lysosomal and aminoacid metabolism, and the development of the fetus and fetal organ maturation. Disturbances of distinct metabolic pathways could therefore be of significant relevance in utero, and may influence intrauterine growth and health. Production of a toxic or energy-deficient intrauterine milieu, modification of the content and function of membranes, or disturbance of the normal expression of intrauterine genes may be responsible for fetal intrauterine compromise and developmental disorders.
The 5 main mechanisms involved in malformative metabolic syndromes are summarized in Table 2. These mechanisms can be isolated or can occur in combination. These mechanisms include energy deprivation, accumulation of toxic metabolites, substrate/nutriments insufficiency, and cell membrane receptor and cell signaling abnormalities.
Mechanisms of malformation of the central nervous system in inborn errors of metabolism.
Brain malformations in Smith-Lemli-Opitz syndrome and in mitochondrial disease are classical examples of the relationship between CNS malformation and inherited neurometabolic disorders.18,19 In Smith-Lemli-Opitz syndrome, neurodevelopmental abnormalities are seen in about one-third of patients, and involve mainly midline and para-midline structures. These structural brain abnormalities include ventriculomegaly, hypoplastic frontal lobes, dysgenesis of the corpus callosum, and, in some patients, cerebellar hypoplasia.18 Formation of midline brain structures occurs between 3 and 12 weeks’ gestation and involves molecular signals that regulate neural tube patterning. Cholesterol serves as an essential co-factor for an important ventral patterning protein, sonic hedgehog. Sonic hedgehog may also play a role in the patterning of dorsal midline structures. Holoprosencephaly, a failure of the forebrain to separate into 2 hemispheres, occurs around the 5th week of gestation. Abnormal sterol levels during embryologic brain development have been shown to disrupt early midline patterning processes, and may represent a mechanism of disease for these malformations. Cholesterol is also involved in myelin structure, membrane lipid raft distribution, activity-dependent synaptic plasticity, and neurosteroid formation.20
The most common developmental brain abnormality associated with mitochondrial disease is complete or partial agenesis of the corpus callosum, a defect of axonal projection. This is commonly found in patients with pyruvate dehydrogenase deficiency and fumarase deficiency. Other anomalies include pachygyria, heterotopias, or pontocerebellar hypoplasia. Pathogenesis of cerebral developmental anomalies in mitochondrial disease involve an energy deficit, accumulation of toxic metabolites such as lactic acid, deficiency of neurotransmitters, or a combination of these factors.19-21
Common brain malformations associated with neurometabolic disorders
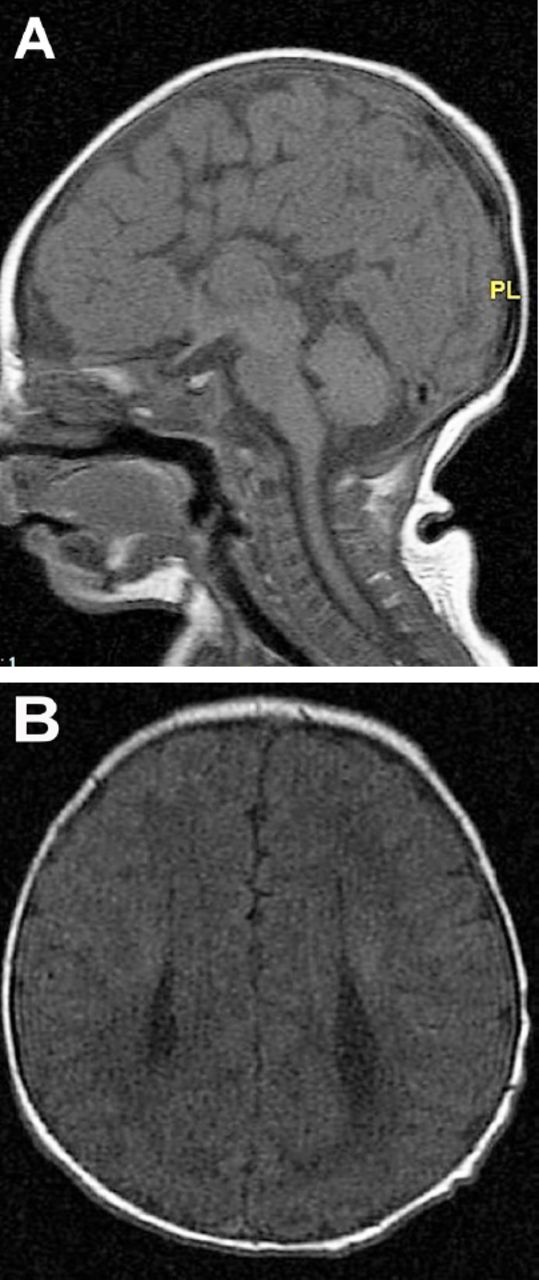
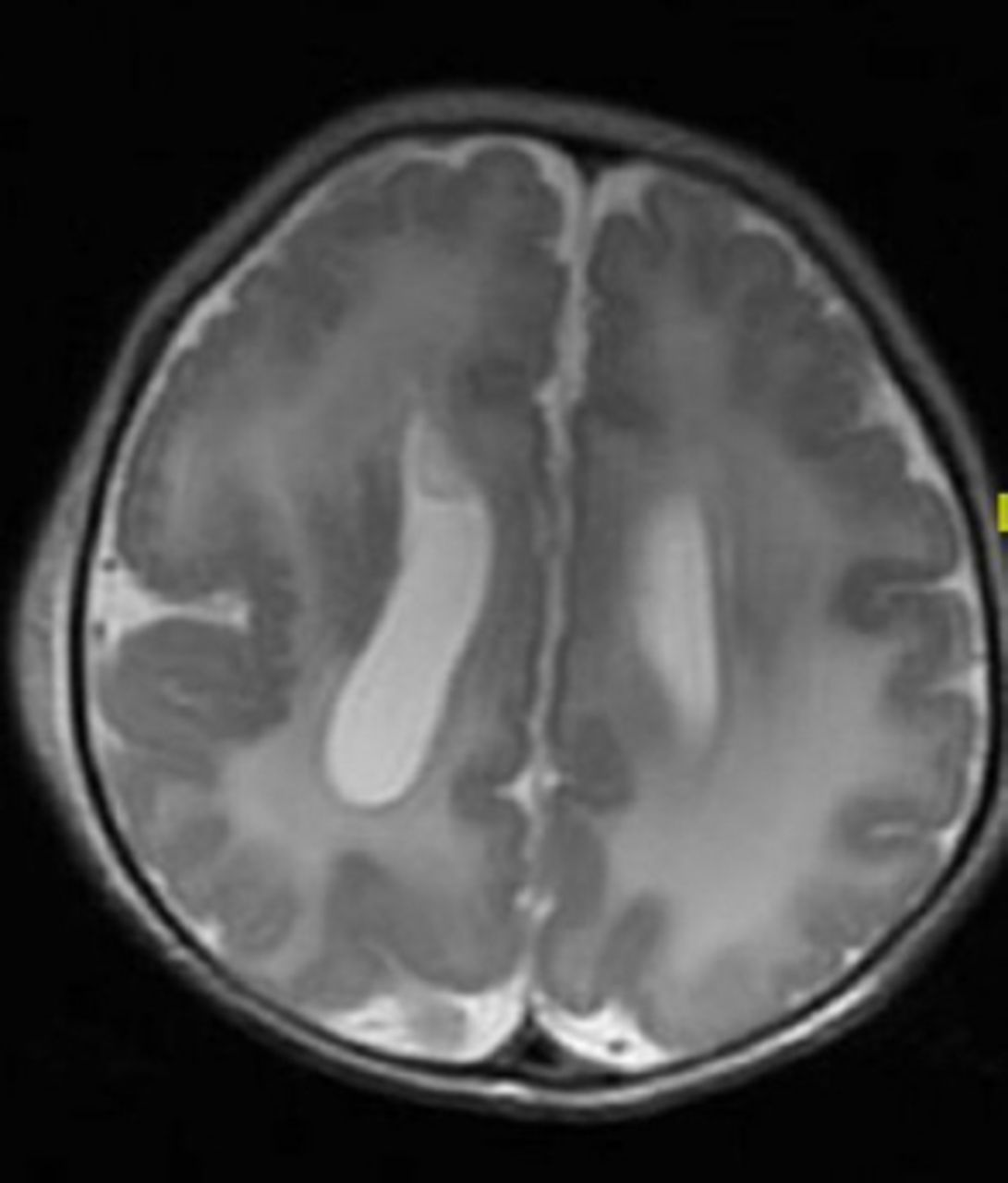
In developed countries, birth defects contribute significantly to infant mortality rates, and are the leading cause of perinatal mortality in some countries.17 Malformations of the CNS are the third commonly observed malformations after cardiovascular system malformations and renal system. CNS malformations are much higher in populations with high consanguinity: 5.6/1000 in Saudi Arabia, compared to 2.3/1000 in Western countries.1 Several case series and case reports highlight the association of brain malformation with inherited neurometabolic disorders. Bamforth reported a 17% association among such patients,7 while Prasad et al. reported a 15% association among such patients.4 Dobyns, drew attention to the association of agenesis of the corpus callosum and cortical malformations in nonketotic hyperglycinemia.10 Recent studies using diffusion-weighted imaging and diffusion tensor imaging in patients with nonketotic hyperglycinemia showed the presence of white matter microstructural alterations, and other non specific brain atrophy. The corpus callosum atrophy can be part of these atrophic changes.22 Nissenkorn et al6 and Gropman et al12 suggested that inborn errors of metabolism play causal roles in the development of cerebral malformations. Congenital malformations of the CNS in the context of inherited neurometabolic disorders include mainly corpus callosum agenesis (Figure 1), midline brain defects, cerebellar/pontocerebellar malformations, and cortical malformations (Figure 2). Table 3 summarizes the common inherited neurometabolic disorders and associated malformations of the CNS.

MRI brain a) sagittal sequence, b) axial sequence. Total agenesis of the corpus callosum in a 7 days old boy with nonketotic hyperglycinemia

MRI brain, axial T2-sequence. Cortical malformation (polymicrogyri) in a patient with Zellweger disease. Note also the abnormal white matter signal.
Common brain malformation associated with neurometabolic disorders.
Practical approach
The recognition of an inherited neurometabolic disorder as the cause for a congenital brain malformation has implications for both the care of the patient, mainly in treatable conditions, and for genetic counseling. Brain malformation can be the dominant feature and mask an underlying inherited neurometabolic disorders.
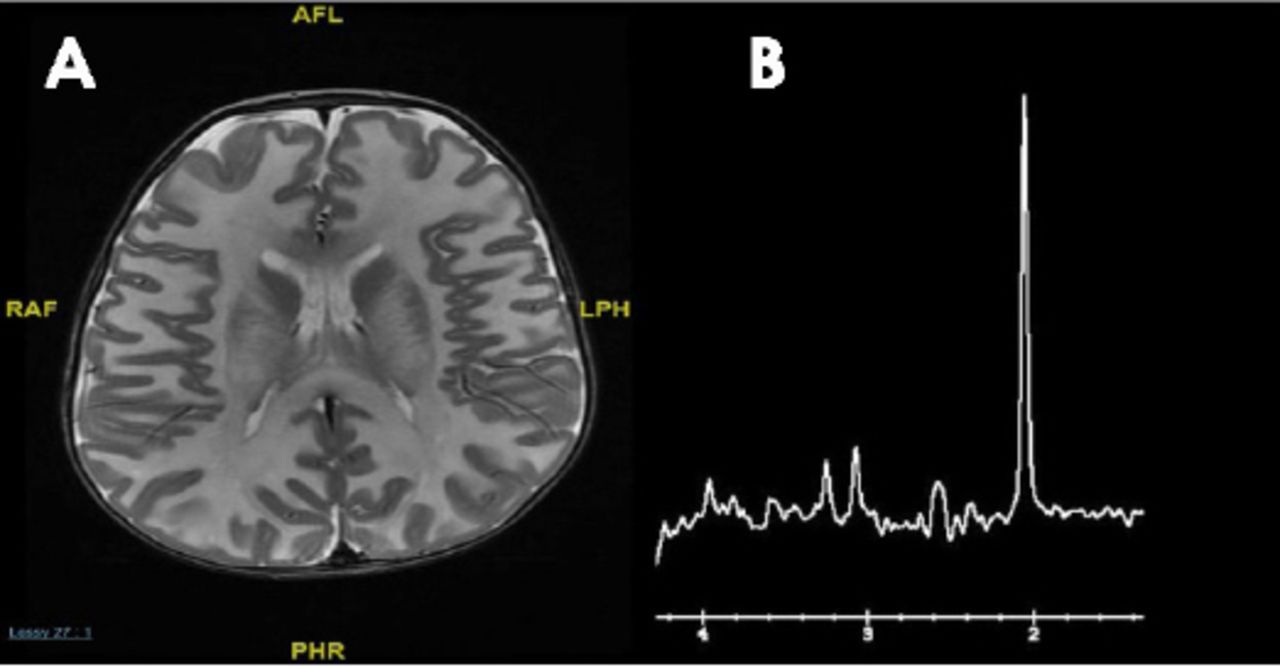
The main challenge is determining which metabolic work-up should be performed following the discovery of a brain malformation by neuroimaging in a patient with developmental disorders, intellectual disability, or epilepsy. Conventional MRI and /MR spectroscopy are essential to delineate the anatomical and metabolic abnormalities respectively, and may give an important clue for an underlying inherited metabolic diseases. The MR spectroscopy is becoming an important adjunct to MRI in the diagnosis of metabolic disorders: specific MRS patterns are found in Canavan disease (elevated n-acetylaspartate) (Figure 3), cerebral creatine deficiency (reduced creatine), and nonketotic hyperglycinemia (presence of glycine), but in most inherited metabolic disease, the MRS findings are not specific.

MRI brain a) axial T2 weighted image of 5 months old patient with Canavan disease demonstrates diffuse abnormal white matter signal; and b) MR spectroscopy showing markedly elevated N-acetylaspartic acid peak.
Three scenarios can be proposed
1. Known phenotype
This is the case of a known, already individualized phenotype: corpus callosum agenesis (Figure 1a & Figure 1b) associated with early onset intractable seizures and burst suppression pattern on the EEG should orient to nonketotic hyperglycinemia. Another common example is the combination of corpus callosum agenesis with congenital lactic acidosis, typical for pyruvate dehydrogenase deficiency. In these cases, the metabolic investigation should target the suspected diagnosis.
2. #phenotype plus: brain malformation associated with systemic manifestations#
The phenotype here includes severe neurological manifestations that are greater than expected with malformations alone or are associated with systemic manifestations such as hepatosplenomegaly or ophthalmologic findings. The work-up should include tandem-mass-spectrometry, urinary organic acid-, and amino acid-profiles, cholesterol, 7-dehydrocholesterol and very long chain fatty acid; measurements of blood lactate, pyruvate, and acylcarnitines profiles. These tests may help to recognize errors of amino acid, galactose metabolism, cholesterol abnormalities, or abnormalities in the energy metabolism.
3. Isolated brain malformation: the role of next generation sequencing
The recent availability of genomic tools that are largely unaffected by phenotyping bias has ushered in a new era of molecular diagnostics and revealed many surprising findings that could not have been predicted solely on clinical grounds. Interestingly, Miller syndrome, a known dysmorphology syndrome for many years, was the first Mendelian disorder to be solved by next generation sequencing, and turned to be an inborn error of pyrimidine metabolism.23 The discovery of genetic defects will link to the metabolic defects. Several gene discoveries expand our understanding of the link between the developmental pathway and the metabolic pathway; neu-laxova syndrome is a typical example which clearly demonstrate the role of disturbance of serine metabolism, and brain abnormalities.24
In conclusion, congenital brain malformations are not uncommon in the context of inherited metabolic disorders, especially those affecting energy metabolism, and the work-up of congenital brain malformations should include inherited neurometabolic disorders. Inherited neurometabolic disorders can lead to disturbances in brain development through multiple mechanisms that include deficits in energy metabolism, critical nutrient deficiency, accumulation of neurotoxic metabolites, abnormality in cell membrane constituents, and interference in cell-to-cell signaling pathways. The anomalies observed include neural tube defects, absent or hypoplastic corpus callosum, midline brain defects, malformations of the cortex, the cerebellum and the brain stem.
Early diagnosis of an underlying inherited neurometabolic disorders is critical for the institution of treatment, which may positively influence clinical outcome, and inform future genetic counseling.
Acknowledgment
The authors gratefully acknowledge Prof.Kalthoum Tlili, Department of Radiology, and Dr. Amal AlAlhashem, Department of Pediatrics, Prince Sultan Military Medical City, Riyadh, Kingdom of Saudi Arabia for the critical review of the manuscript, and their valuable comments and advice.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received October 12, 2017.
- Accepted December 27, 2017.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.