


Clinical Presentation
A 6-month-old girl was admitted with a 2-month history of irritability, unexplained intermittent fever, developmental regression, and myoclonic seizures. She is the product of first-degree consanguinity. The family history was remarkable for one sister who died at the age of 2 years with a similar neurological condition. Neurological examination showed excessive irritability, hypertonicity of the 4 limbs, hyperreflexia, and bilateral extensor plantar reflex. Systematic examination was unremarkable. A brain MRI was performed and is shown in Figures 1 & 2.

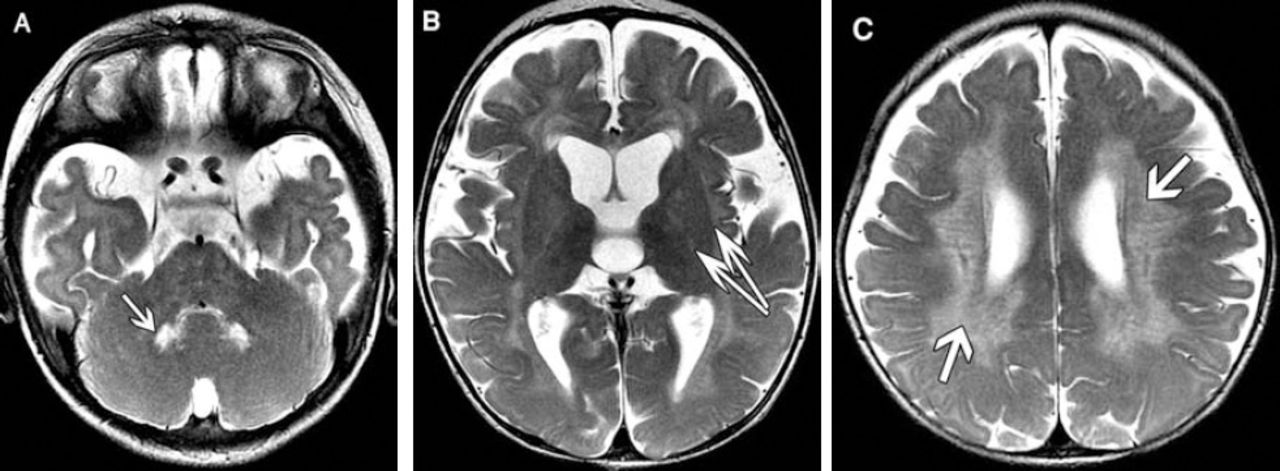
Axial T2-weighted patient brain MRI.

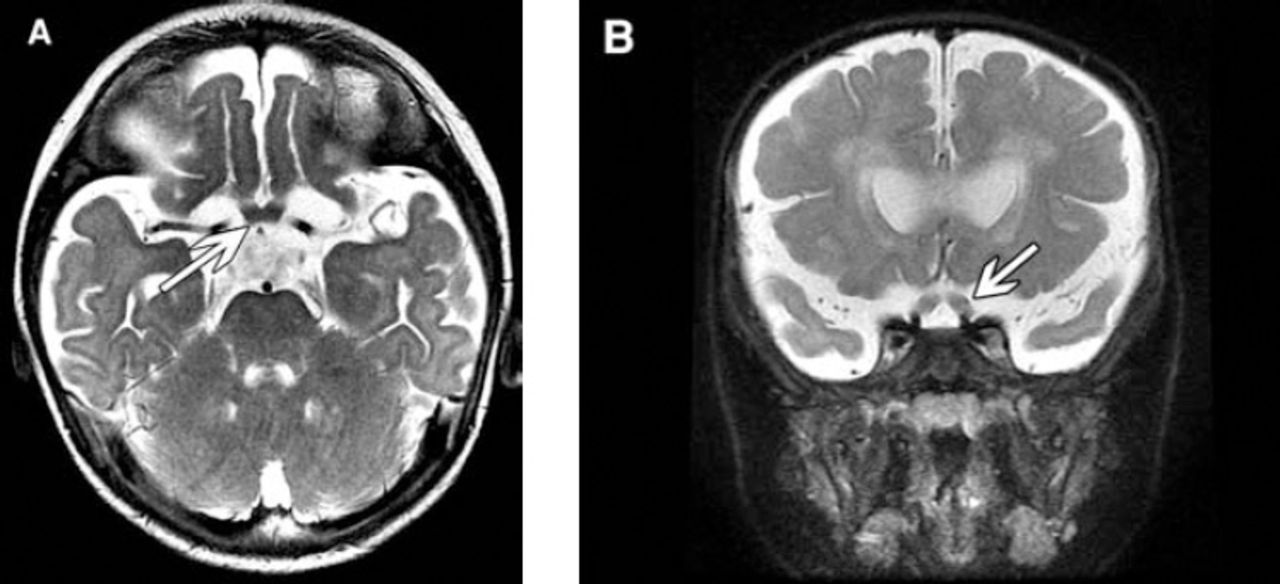
Axial and coronal T2-weighted patient brain MRI.
Questions
Answers & Discussion
MRI findings. Figure 1: Axial T2-weighted images show A) hyperintense signal in deep cerebellar nuclei (arrow) bilaterally, B) in the bilateral posterior limb of the internal and external capsules, and C) bilateral symmetrical confluent hyperintense signals in the centrum semiovale and periventricular region sparing subcortical U fibers. Tubular linear areas of hypointensity are noted within the demyelinating bright areas extending from the periventricular region toward the cortex giving a tigroid pattern (stripe sign). Figure 2: Axial and coronal T2-weighted images (A & B) shows marked thickening of the optic nerves (arrow) and chiasm.
Diagnosis. Based on the clinical presentation and neuroimaging findings, the most likely diagnosis is leukodystrophy type Krabbe disease.
Confirmatory diagnosis of Krabbe disease requires enzymatic assay: b-galactocerebrosidase activity in peripheral blood leukocytes or skin fibroblasts. In our patient b-galactocerebrosidase enzyme activity was deficient. Genetic study showed mutations in the galactocerebrosidase gene.
Krabbe disease is an autosomal recessive lysosomal storage disease caused by mutations in the galactocerebrosidase gene leading to deficiency of the enzyme b-galactocerebrosidase, a key enzyme in the catabolism of myelin-enriched sphingolipids. Accumulation of globoid cells results in optic nerve enlargement.1 Clinically, the disease manifests early in infancy in 70-80% of patients. Infants usually present with spastic quadriparesis, tonic spasms, extreme irritability, inconsolable crying, progressive optic atrophy, peripheral neuropathy, and death usually due to severe respiratory infection at around the second year of life. The juvenile and adult forms are characterized by insidious visual compromise, cognitive deterioration, and gait disorders.1
The MRI abnormalities differed among phenotypes.2 Early infantile patients had a predominance of increased intensity in the dentate/cerebellar white matter as well as changes in the deep cerebral white matter. Later onset patients did not demonstrate involvement in the dentate/cerebellar white matter, but had extensive involvement of the deep cerebral white matter, parieto-occipital region, and posterior corpus callosum. Late infantile patients exhibited a mixed pattern. Adolescent/adult patients demonstrated isolated corticospinal tract involvement. The brain MRI may also show enlargement of the optic nerve and optic chiasma. Spinal MRI with contrast shows lumbosacral nerve root contrast enhancement. The differential diagnosis of optic nerve enlargement includes glioma, nerve sheath meningioma, dural ectasia, and neurofibromatosis type I.3
The diagnosis of Krabbe disease is challenging because of the large number of leukodystrophies, as well as the similarities between their clinical and imaging findings. Specific neuroimaging findings may help to establish this diagnosis. The detection of optic nerve enlargement, or lumbosacral nerve root contrast enhancement not found in other types of leukodystrophies, is an auxiliary tool in the differential diagnosis of KD.
Treatment of Krabbe disease is mainly symptomatic. At present, the only clinical treatment is hematopoietic cell transplantation. It is beneficial if performed before the onset of symptoms, but its efficacy in correcting the severe neurological disease is variable. Gene therapy is a promising alternative.1
Footnotes
Notice: Authors are encouraged to submit clinical images for possible publication in the Journal. These may be in any field of Clinical Neurosciences, and should approximately follow the format used here. Please address any submissions to the Assistant Editor, Neurosciences Journal, Prince Sultan Military Medical City, PO Box 7897, Riyadh 11159, Kingdom of Saudi Arabia. E-mail: sdouglas{at}psmmc.med.sa
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.