


The congenital cranial dysinnervation disorders (CCDDs) encompass most congenital, static restrictions of ocular motility, often associated with ptosis, and retraction of the globe.1 The CCDDs can be monogenic or chromosomal in origin. Although chromosomal copy number variations (CNVs) has been reported in patients with syndromic CCDDs,2 but there are still patients for whom the genetic basis has not been identified yet. In this report, we describe the evaluation of a girl with CCDD and other congenital abnormalities by high-resolution array-comparative genomic hybridization (array CGH), which revealed a microdeletion in chromosome 6.
A 6-year-old girl with strabismus and multiple congenital anomalies was referred for genetic evaluation. She had a history of mild pulmonary valve stenosis and neonatal seizures that did not recur after the neonatal period. Esotropia, ptosis on the right, polydactyly of both hands, and cleft palate were noted at birth. She had a cleft palate repair, strabismus surgery (uncomplicated bilateral medical rectus muscle recessions for esotropia), and ptosis surgery (uncomplicated right frontalis sling) during early life. She had no other known general medical problems. Examination revealed a pleasant and alert girl who appeared small for her age (head circumference 48 cm, height 90 cm, weight 11.95 kg). Ophthalmologic examination was significant for ptosis on the right and an alternating esotropia of approximately 20 prism diopters. She had bilateral complete abduction defects (Figure 1) with retraction of each globe on abduction consistent with co-contraction of the medial and lateral rectus muscles. She was also unable to supraduct either eye. Adduction, infraduction, and pupillary examination were normal. The ptotic lid on the right eye elevated during adduction. Cycloplegic refraction revealed mild myopia with an otherwise normal posterior pole in both eyes. Clinically, she did not have any skeletal abnormalities other than polydactyly, and a plain film skeletal survey did not show unusually long tubular bones, tall vertebral bodies, or other significant bony abnormalities. A head CT performed soon after birth was reported as normal, but subsequent neuroimaging was not performed because she was cognitively normal. The patient’s parents were not related, and she had 2 siblings, a normal older brother and a sister who had decreased vision by history (Figure 1E).
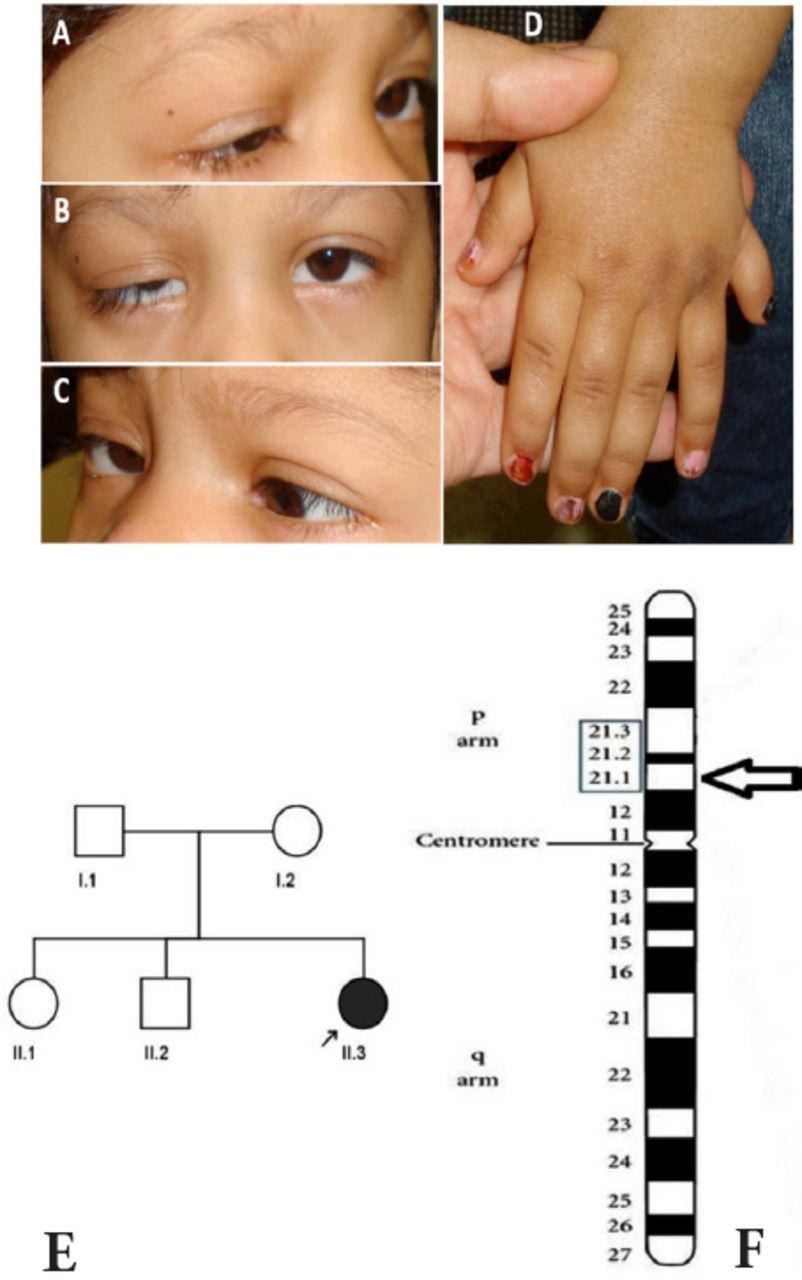
Clinical and genetic findings: A) on attempted right gaze, abduction is absent and right ptosis is evident; B) in primary position, there is a small angle esotropia and right ptosis is evident; C) abduction is also absent on attempted left gaze, and the right lid elevates; D) polydactyly of the left hand is illustrated; E) family pedigree (arrow indicates proband); and F) Ideogram of chromosome 6 with the deletion location on the cytogenetic band marked by arrow head.
The study adhered to the principles of Helsinki Declaration. The patient and 5 family members were recruited under an institutional review board protocol approved at the King Khalid Eye Specialist Hospital, Riyadh, Kingdom of Saudi Arabia (KSA) (0424-P). The informed consents were obtained. The patient was examined, and her chart was reviewed at the Ophthalmic Genetics Laboratory, King Abdulaziz University Hospital, King Saud University, Riyadh, KSA from May to August 2013. Candidate genes associated with syndromic DRS (HOXA1, SALL4, CHN1, TUBB3, and KIF21A) were screened. The complete coding regions of the SALL4, CHN1, HOXA1, and TUBB3 genes and exons 8, 20, and 21 considered hotspot for mutations in the KIF21A gene were sequenced according to protocols described previously.2 The Affymetrix Cytogenetics Whole-Genome 2.7M array (Affymetrix Inc., Santa Clara, CA, USA) was used to detect chromosomal aberrations across the entire genome as described elsewhere.2 The data analysis was carried out using Affymetrix® Chromosome Analysis Suite v1.2 (ChAS) Software (Affymetrix Inc. CA, USA). A CNV was considered potentially pathologic if it satisfied all of the following preliminary devised criteria: 1) it was not reported in the Database of Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home) among normal controls; 2) it was not present in at least 215 healthy controls of similar ethnicity; 3) it segregated with the phenotype and was not present in unaffected family members; and 4) it included an area of the genome encompassing one or more functional gene(s). The threshold for gain or loss was adjusted to 10 Kb. We used the National Center for Biotechnology Information Human Genome Assembly Build 35. A semi-quantitative polymerase chain reaction (PCR) method was performed on the 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) to confirm the array CGH findings as described earlier.2
Sequencing results did not show any variations in the screened regions of SALL4, CHN1, HOXA1, TUBB3, and KIF21A genes. However, array CGH results documented a deletion in chromosome 6 extending from 43,002,148 to 43,018,858 (total size 16.7 Kb) at the 6p21.1 cytogenetic band (Figure 1F). The copy number state was equal to 1, indicating that this deletion was likely to be heterozygous. The confidence value calculated by ChAS software was 88% with a marker count of 20 spanning the deleted area. This deletion was likely to be de novo and segregated with the syndrome described here because it was not detected in the proband’s mother or father, or 215 unrelated healthy individuals of similar ethnicity. The deletion was confirmed by semi-quantitative PCR. The mean (standard deviation) of 3 separate readings of fluorescence peak area was 689.4 (11.8) for the proband, 901.7 (25.4) for the mother, 917.9 (28.6) for the father, and 965.2 (18.2) for a normal female control of similar ethnicity. This 16.7 Kb deletion region encompassed only one functional gene, cullin 7 (Gene Symbol CUL7, NC_000006.11).
The proband described here had bilateral limitation of abduction and supraduction with retraction of either globe on attempted abduction, ptosis on the right, cleft palate, polydactyly of both hands, pulmonary valve stenosis, and neonatal seizures. Her parents were not related and no other family member had strabismus or any other obvious congenital anomaly. Thus, this patient qualifies for the diagnosis of syndromic CCDD. She did not fit the classical phenotype for previously recognized monogenic CCDD syndromes,1 and she also did not have mutations in HOXA1, SALL4, CHN1, TUBB3, and KIF21A genes. Thus, a chromosomal anomaly seemed to be a possible genetic cause of her syndrome. High resolution array CGH studies documented only one small chromosomal anomaly. This unique CNV was a 16.7 Kb heterozygous deletion at the 6p21.1 cytogenetic band and involved only the CUL7 gene.
The CUL7 gene encodes the cullin 7 protein, which is a component of the E3 ubiquitin ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of unwanted target proteins. The ubiquitin-proteosome system regulates cell division and growth by antagonizing p53 function. Autosomal recessive mutations in the CUL7 gene sometimes cause the rare 3-M syndrome,3 which causes intrauterine growth retardation in humans with resultant short stature (dwarfism), unusual facial features (such as, triangle-shaped face with a pointed chin, frontal bossing, a hypoplastic mid-face, a short upturned nose with a fleshy tip), and skeletal abnormalities. The patient reported here did not have most of the features described in patients with the 3-M syndrome including the facial dysmorphism or skeletal abnormalities that are required for its definitive clinical diagnosis. However, she did have a cleft palate and polydactyly, dysmorphic features that are reminiscent of the facial and orthopedic abnormalities of the 3-M syndrome. A variant similar to 3-M syndrome, called the Yakut short stature syndrome with novel CUL7 mutation has also been identified in an isolated population in Siberia.4 Infants affected with this disorder exhibit severe asphyxia and respiratory problems that can be life-threatening.4 A similar deletion involving chromosome 6p21.2-p12.3 was described by Chen et al5 in an 8 year-old girl but with different phenotypic characteristics including cleidocranial dysplasia, mild intellectual disability, developmental delay, and poor wound healing. These individuals do not resemble the proband.
To summarize, we speculate that the microdeletion in the CUL7 gene may be responsible for the development of CCDD, cleft palate, and other congenital abnormalities in this patient. However, development of normal ocular motility is a complex process that is not yet fully understood. Therefore, the role of the hemizygous CUL7 gene in causing CCDD and the other described features cannot be proven unequivocally from this one patient, but a convincing link may be established in the future if these observations are replicated in more individuals.
It may be possible that other genetic abnormalities not tested for by the technology employed here may be responsible for the patient’s syndrome; or that the patient has a chromosomal deletion smaller than 10 Kb, which is below the resolution of current technology; or that an unidentified mutated gene might not have been symptomatic in apparently unaffected family members. Finally, this syndrome may be the result of unrecognized environmental or epigenetic factors. These issues can only be resolved after more patients have been reported with small deletions in the same chromosomal region.
Acknowledgments
The authors are highly indebted to the Glaucoma Research Chair at King Saud University, Riyadh, Kingdom of Saudi Arabia, for allowing the use of their laboratory facilities for this work.
Footnotes
Disclosure
This study was funded by the National Program for Science and Technology (Grant # 12-MED2621-02), King Saud University, Riyadh, Kingdom of Saudi Arabia.
- Received February 3, 2015.
- Accepted August 26, 2015.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.