


Abstract
Objectives: To identify genetic variation involved in primary microcephaly.
Methods: In present study we identified 4 generation Saudi family showing primary microcephaly. We performed whole exome sequencing along with Sanger sequencing to find the genetic defect in this family. This study was conducted in King Abdulaziz University started from 2016 and the results presented in this manuscript are from one of the family.
Results: Two novel missense variants (c.982G>A and c.1273T>A) were identified in heterozygous state in exon 8 of MCPH1 gene. The detected missense variants cause a tyrosine to asparagine substitution of residue 425 and a valine to isoleucine substitution at residue 310. MCPH1 gene encodes a DNA damage response protein. The encoded protein play a role in G2/M DNA damage checkpoint arrest via maintenance of inhibitory phosphorylation of cyclin-dependent kinase 1. The respective mutation was ruled out in 100 control samples.
Conclusion: We found novel compound heterozygous mutation in Saudi family that will help to build database for genetic mutations in population and pave way to devise strategies to tackle such disorders in future.
Primary Microcephaly (MCPH, OMIM 251200) is referred to as the congenital genetic neurodevelopmental defect that affects the brain size and intelligence.1 Typically the brain size or occipital frontal circumference (OFC) at the time of birth is less than four standard deviations (-4 SD) below the age, sex and ethnicity matched control group.2 Most of the primary microcephaly patients present with normal weight, height and appearance along with normal banding pattern of chromosomal analysis and brain scan result.3,4 Few exceptions of this rule is observed in the patients with underlying mutations in microcephalin gene, who are shown to have short stature along with abnormal chromosomal analysis results.5 Studies on brain growth also revealed that primary microcephaly is due to the abnormal developmental process and not because of the degeneration or regression of the neuronal tissues.6
The incidences of primary microcephaly is much higher in the populations where cousin marriages and higher birth rate is found like in Middle East and South Asia and lower in Caucasians where consanguinity is lower.1 There are 18 OMIM genes associated with primary microcephaly are reported so far. The genes include MCPH1 (*607117), WDR62 (*613583), CDK5RAP2 (*608201), CASC5 (*609173), ASPM (*605181), CENPJ (*609279), STIL (*181590), CEP135 (*611423), CEP152 (*613529), ZNF335 (*610827), PHC1 (*602978), CDK6 (*603368), CENPR (*616051), SASS6 (*616402), MFSD2A (*616486), ANKLE2 (*616681), CIT (*617090) and WDFY3 (*617520).7
MCPH1 was the first gene reported in primary microcephaly.8 It is located at the chromosome position 8p23 with genomic size of 241,905 and has 14 exons encodes a protein of 835 amino acids. This gene has three already known isoforms and 8032 base pairs of open reading frame.9 The first mutation in MCPH1 gene was reported in Pakistani families.9 To date 27 mutations have been identified in this gene. The animal models were also used to study the disrupted MCPH1 revealed unregulated mitotic chromosome condensation and reduced head circumference.10,11
Recently we have studied novel mutation in PGAP2,12 STAMBP13 and WDR627 genes cause primary microcephaly in a consanguineous Saudi families. In this study we had identified another Saudi pedigree with 2 primary cases and the aim was to identify the molecular basis i.e. genetic variants in the responsible gene underlying this disorder. We identified 2 novel missense variants (c.982G>A and c.1273T>A) in the heterozygous state in exon 8 of MCPH1 gene to leading to disease further explain the role of MCPH1 gene in Saudi population.
Methods
We designed a retrospective study with detailed family history of disease with suspected risk and recruited a consanguineous 4 generation Saudi family with 2 affected members with primary microcephaly. Pedigree was carefully constructed by interviewing the elders of the family as shown in Figure 1. This family was included in this study because the phenotypes of the patients was very clear for primary microcephaly and the patients were laying under the inclusion criteria as set for this study. We exclude the families those have not clear phenotype of primary microcephaly or without having the detailed phenotype and clinical information. Blood samples of both parents and affected children were taken for DNA extraction and further molecular analysis. Moreover blood sample was also taken from 100 healthy unrelated control persons. This study was done followed by Helsinki declaration and approved by the ethical committee of CEGMR. Informed consent was taken prior to the study from all participants.
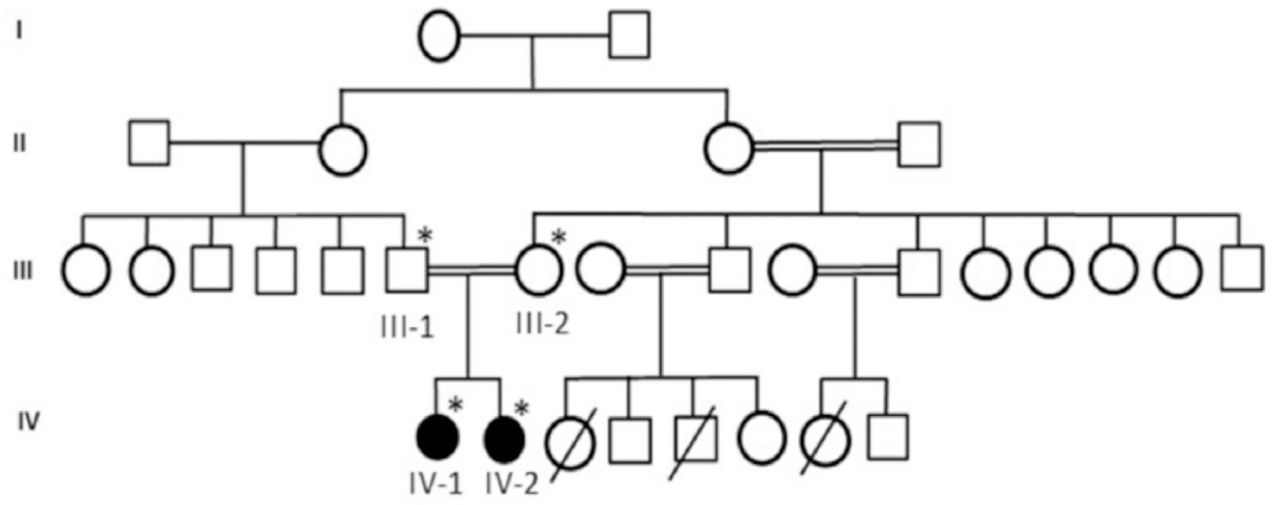
A consanguineous family of primary microcephaly from Saudi Arabia showing the disease phenotype segregating in an autosomal recessive manner. The samples available for genetic testing were marked with asterisks.
First patient is a 10-year-old girl. At the time of blood sampling her head circumference was 44 cm (-5SD below the same age and sex). She was intellectually disabled. She can not speak mother language properly, nor able to express her feeling well. She had typical feature of primary microcephaly like sloppy forehead but with no other neurological finding, such as spasticity, seizures, or progressive cognitive decline.
The second patient is a 5-year-old girl. Her head circumference measured 41 cm (-6SD below the same age and gender). She was mentally retarded by birth with primary microcephaly. Her facial features are like others previously reported with primary microcephaly like sloppy forehead etc. She was also not able to speak properly and not able to express her feelings well.
To find out the pathogenic mutation that may be the cause of disease whole exome sequencing was performed for both affected individuals (IV-I and IV-2) of the family. The samples for exome sequencing were prepared according to an Agilent SureSelect Target Enrichment Kit preparation guide (SureSelect_v6 Agilent USA) and exome sequencing was carried out using Illumina HiSeq 2000/2500. Further, the libraries were sequenced using HiSeq 2000/2500 sequencer from Illumina. The variants obtained were filtered based on different parameters such as the quality, frequency, effect of protein, its genomic position, pathogenicity and known associations with the disease. Different bioinformatics tools were used to find the any of the causative variant co-segregating with the disease of microcephaly as an autosomal recessive manner. Further we used Laser gene Genomic Suite v. 12 (DNASTAR, Madison, WI, USA) and ArrayStar v. 12 (Rockville, MD, USA) to identify variant alleles based on dbSNP. The acquired FASTQ files were used to aligned the BWA Aligner (http://bio-bwa.sourceforge.net/) and hg19 (NCBI build GRCh37) by using SeqMan NGen 12 (DNASTAR). The sequence reads were mapped against the human reference sequence hg19 (http://genome.ucsc.edu/) and compared with 1000 Genomes Project databases (http://www.1000genomes.org/data) and sequences in the dbSNP (http://www.ncbi.nlm.nih.gov/snp/) available online.
Subsequently to confirm the mutation we found in whole exome sequencing, Sanger sequencing was perform in father and mother of children as well as in 100 control individuals. MCPH1 gene were amplified by polymerase chain reaction (PCR). Purified PCR products were subjected to cycle sequencing reactions by using BigDye Terminator V3.1 Cycle Sequencing kit to detect any mutation. To confirm the mutation in patients and family, we did Sanger sequencing using Applied Biosystems 3500 (CA, USA) Sequencer. We also sequenced this DNA variant in unrelated 100 healthy control people to confirm the variant as pathogenic.
Insilico analysis and functional prediction of these mutations were analyzed using the online prediction software tools that includes PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) PROVEAN/SIFT (http://provean.jcvi.org/) and PhastCons (http://compgen.cshl.edu/phast/) etc.
Results
Whole Exome and Sanger Sequencing
Whole exome sequencing revealed 2 compound heterozygous missense mutations in exon 8 of MCPH1 gene in both affected individuals where guanine at position 982 is converted to adenine (c.982G>A) and thymine at position 1273 is converted to adenine (c.1273T>A). These missense variants cause a tyrosine to asparagine substitution of residue 425 and a valine to isoleucine substitution at residue 310. Furthermore, the parents were sequenced by using Sanger sequencing and found one heterozygous missense mutation at each position. Whole exome sequencing revealed pathogenic compound heterozygous mutation in where (c.982G>A and c.1273T>A) in exon 8 of MCPH1 gene.
Sanger sequencing
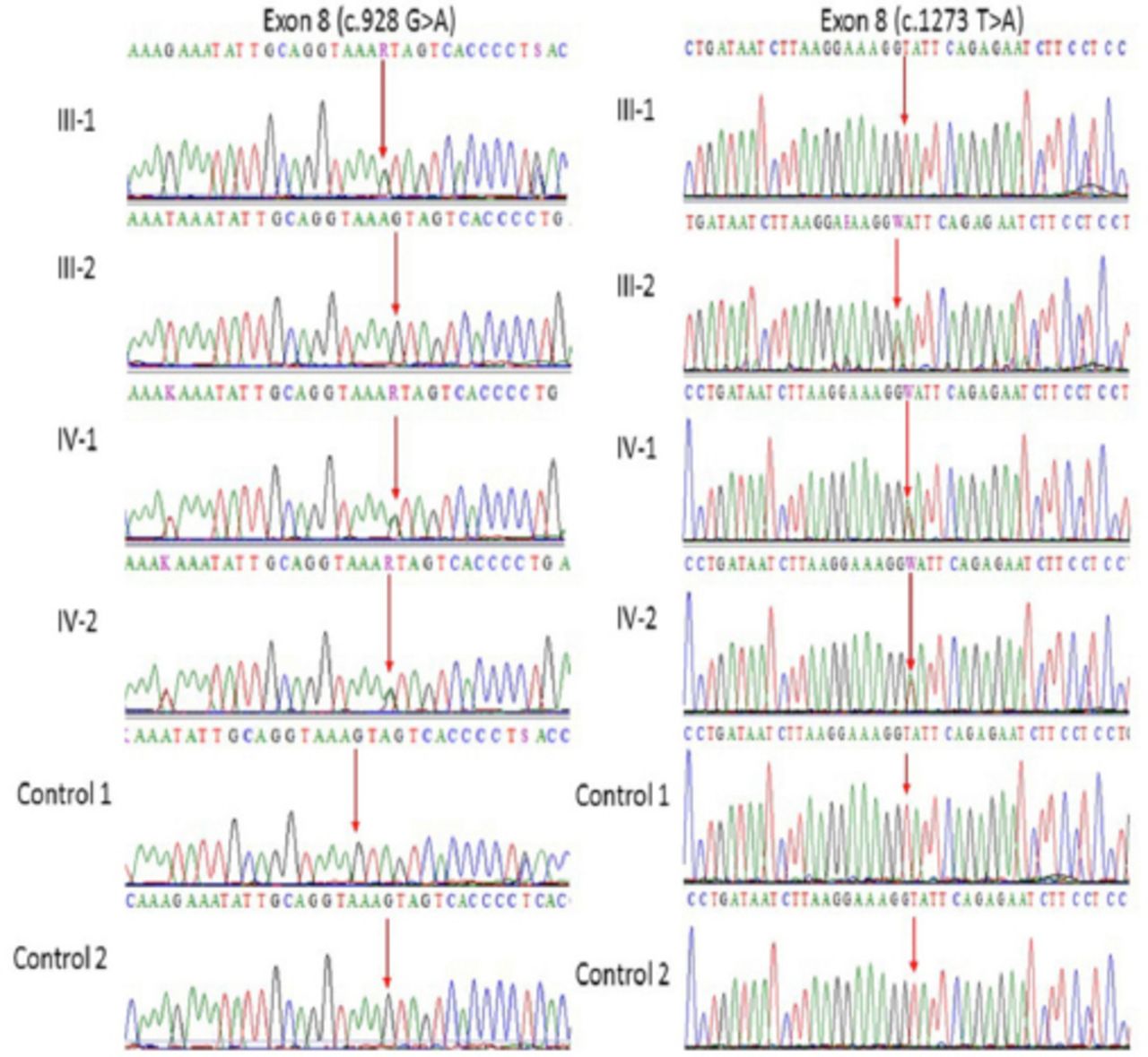
Our Sanger sequencing results further validated a compound heterozygous mutation in MCPH1 gene where at 982 (c.982G>A) and at position 1273 (c.1273T>A) in exon 8 of the both affected IV-1, and IV-2 proband whereas the one parent was heterozygous at one position while other was heterozygous at other position as shown in (Figure 2). This mutation further validated in 100 unrelated healthy persons, but no one has this sequence variation. Both the parents of the affected members were heterozygous at different positions, which also confirms the compound heterozygosity in this family.

Representative electropherogram after Sanger sequence analysis confirm the compound heterozygous mutation in this family, a and b (father III-1 is heterozygous at position c.982G>A and mother III-2 is homozygous whereas at position c.1273T>A father is homozygous while mother is heterozygous) while (IV-1 and IV-2) are affected children showing heterozygosity for both (c.982G>A and c.1273T>A) in exon 8 of MCPH1 gene.
Insilico analysis
Bioinformatics and functional prediction of mutations for deleterious effects were performed using the online In silico prediction software package PolyPhen-2, PROVEAN/SIFT, and PhastCons as shown in Table 1. Furthermore, in the Greater Middle East (GME) variome minor allele frequency was 0.00 in the database. Moreover, SIFT 0.12, PolyPhen 0.7, PhyloP (phyloP46way_placental) and MutationTester (2.0) predicted this variation as disease causing mutation. This mutations was absent in the Human Gene Mutation database (HGMD, www.hgmd.cf.ac.uk/) and Online Mendelian Inheritance in Man (MIM/OMIM). 1000 genome (http://www.internationalgenome.org/) and The Exome Aggregation Consortium (ExAc) (Version 0.3.1) (http://exac.broadinstitute.org/) data base. All of the software’s predicted this mutation to be disease causing and lethal for overall proper functioning of the protein.
In silico tools used for prediction of pathogenicity of missense variants.
Discussion
Mutations in MCPH1 gene have been related with primary autosomal recessive microcephaly 1 and premature chromosome condensation syndrome. The 2 missense variants were identified in heterozygous state in the MCPH1 gene in this study. MCPH1 encodes a regulator of chromosome condensation. The gene has been suggested to play a role in the regulation of the size and control the neurogenesis of the cerebral cortex.9,14
Previously autosomal recessive mutations in this gene are known to cause Primary microcephaly 1 that in which the phenotype of the patient head circumference less than 3 standard deviations (SD) below the age- and sex-related mean, present at birth.2 Microcephaly is a disorder of small brain growth and individuals with microcephaly have small brains size, mild to severe mental retardation, language skills and delayed speech, though some rare individuals with mild microcephaly and normal intelligence have also been reported. Furthermore some other clinical features may include short stature or mild seizures. Motor skills, such as standing, sitting, and walking, may also be mildly delayed. Based on the referral notes for this patient, primary microcephaly 1 could be of relevance for the reported microcephaly. In addition, MCPH1-related primary microcephaly is typically not associated with dysmorphic features (not detailed for this patient). The detected missense variants cause a tyrosine to asparagine substitution of residue 425 and a valine to isoleucine substitution at residue 310. These variants have not been described in literature yet, so caution is warranted regarding possible pathogenicity. Although these variants are quite rare in the general population (no homozygotes for these variants in gnomAD) and the missense changes are clearly predicted to be damaging by in silico tools. Functional studies would be required to determine the effect of the here detected missense change on MCPH1 structure and function. Of interest, previously reported pathogenic mutations in MCPH1 include truncating (nonsense, frame shift), missense mutations and larger deletions.
More detailed clinical information could give more insight into the relevance of these MCPH1 variants. Both missense variants detected here were classified as variants of unknown clinical significance according to the most recent ACMG guidelines.15
In conclusion in this study we report a novel compound heterozygous mutation in MCPH1 gene causing primary microcephaly. In future for further analysis of these variants more families should be tested and animal models should be made and studied for better understanding and management of this disease.
Acknowledgment.
The authors would like to thank King Abdulaziz City for Science and Technology, Science and technology Unit, for technical and financial support.
Footnotes
Disclosure. This project was funded by the King Abdulaziz City for Science and Technology (KACST), Riyadh, Kingdom of Saudi Arabia, under grant number APR-34-13.
- Received February 25, 2018.
- Accepted September 12, 2018.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.