


Abstract
Dyk-Davidoff-Masson Syndrome (DDMS) is one of the rare neurological conditions attributed to the development of drug-resistant epilepsy (DRE). The DDMS condition is characterized by cerebral hemisphere asymmetry, where atrophy occurs on one side of the brain and clinically manifests as hemiparesis, seizure disorder, mental retardation, and facial asymmetry. In addition, the condition has various perinatal or postnatal etiologies. Herein, we report the case of a 29-year-old right-handed male with Dyke-Davidoff Masson syndrome and mild right-side weakness. The patient experiences attacks of seizures with stiffness in his right arm and right leg, sometimes experiencing agitation and abnormal movement of the body parts. The MRI of the brain showed asymmetry with atrophic changes involving the left hippocampus, consisting of mesial temporal sclerosis. Additionally, the results showed the presence of gyral hyperintensities over the left parietal region. Therefore, the patient’s case is reported with a literature review to support it.
Dyke-Davidoff-Masson syndrome (DDMS) is a condition that is characterized by hemiatrophy of the cerebral hemisphere and contralateral hemiparesis, seizures, mental retardation, facial asymmetry, and schizophrenia.1 Hyper-pneumatization of the frontal sinuses, cerebral hemiatrophy, and calvarial thickening are the classical findings noted if an insult to the brain occurs before three years of age.2 Intractable seizures remain the primary concern of the disease, for which drug therapy is insufficient in most cases and a surgical approach is necessary.
Case Report
Patient information and clinical findings
A 29-year-old right-handed man, has been known to have epilepsy since the age of 15. The patient presented with frequent attacks of seizures accompanied by stiffness in both the right hand and leg. He experienced an aura accompanied by chest tightness reported tonic-clonic seizures followed by agitations and sudden abnormal movement of body parts such as hands. He did not provide a significant family and birth history, making it hard to determine whether the condition developed after birth or through hereditary factors (Figure 1).
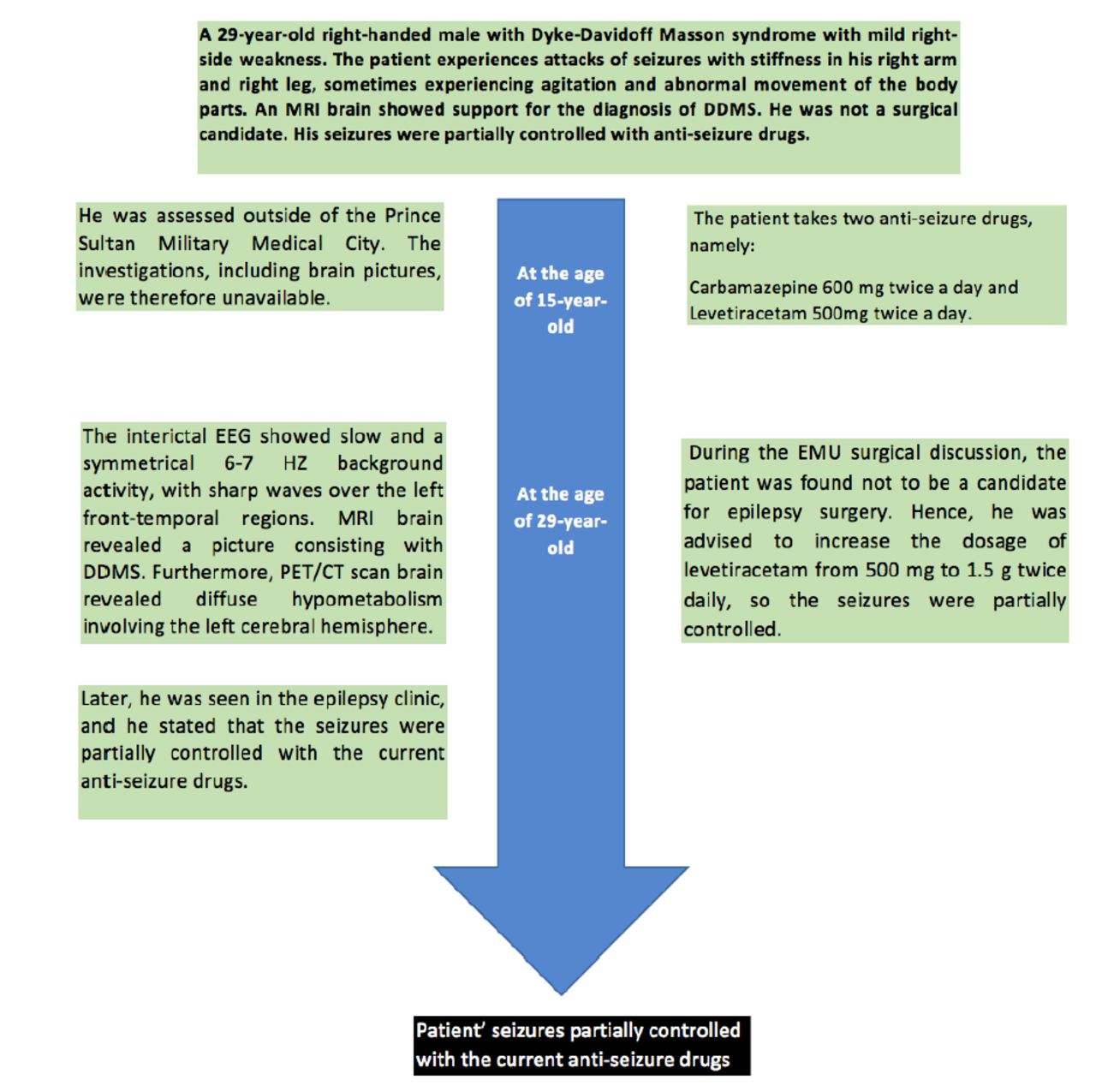
- A timeline showing the course of the patient during follow-up and the outcome.
On physical examination, the cranial nerve examination was unremarkable, and no neurocutaneous manifestations were observed. However, he scored 20/30 on the Mini-Mental Status Examination and had a right-sided spastic hemiparesis with brisk tendon reflexes and an extensor plantar response.
Diagnostic assessment
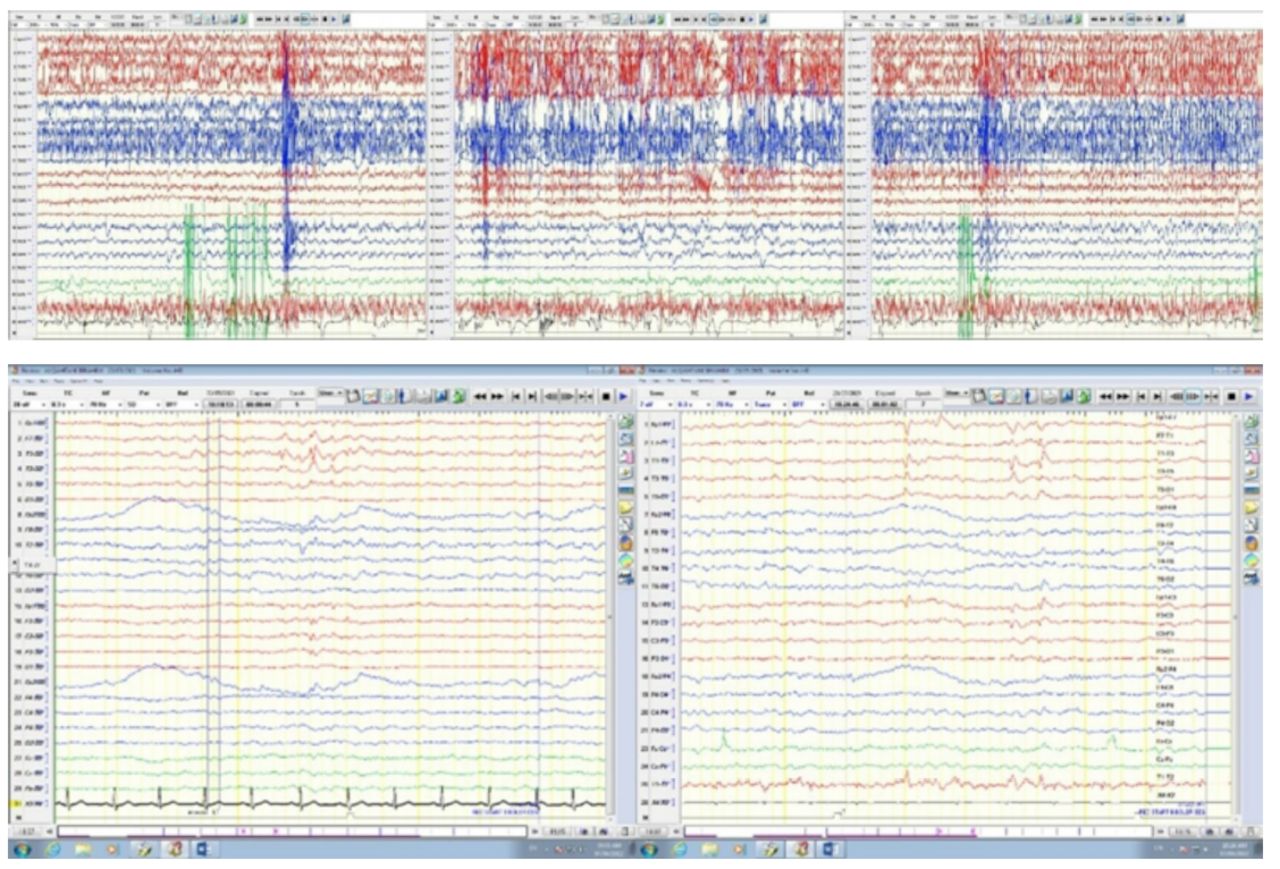
The patient was admitted to an epilepsy monitoring unit for 8 days. The interictal EEG showed slow and a symmetrical 6-7 Hz background activity, with its suppression being over the left hemisphere in the left frontal region. The left temporal and frontal regions were found to have sharp waves, while a slow, intermittent activity was realized over the right frontal-temporal regions (Figure 2A).

- The EEG Shows A) Ictal EEG shows a diffuse myogenic artifact, which is followed by a buildup of rhythmic theta activity in the right parasagittal region, evolving in frequency and amplitude. B) The interictal EEG showed a symmetrical 6-7 HZ background activity, with its suppression being over the left hemisphere in the left frontal region. Sharp waves in the left temporal and frontal regions were observed.
During the seizures, the patient was observed to show sudden onset agitation, screaming, and rubbing of hands and face, followed by holding the right hand on the chest and rocking movements while assuming distal dystonic posturing of both the right upper and lower limbs. The event was recorded at a frequency of 3 times in 8 days. Bicycling movement was also recorded, which was followed by automatic, left-handed behavior moving in the form of genital manipulation. Electrographically, results indicated diffuse myogenic artifacts, followed by rhythmical activity in the right parasagittal region (Figure 2B).
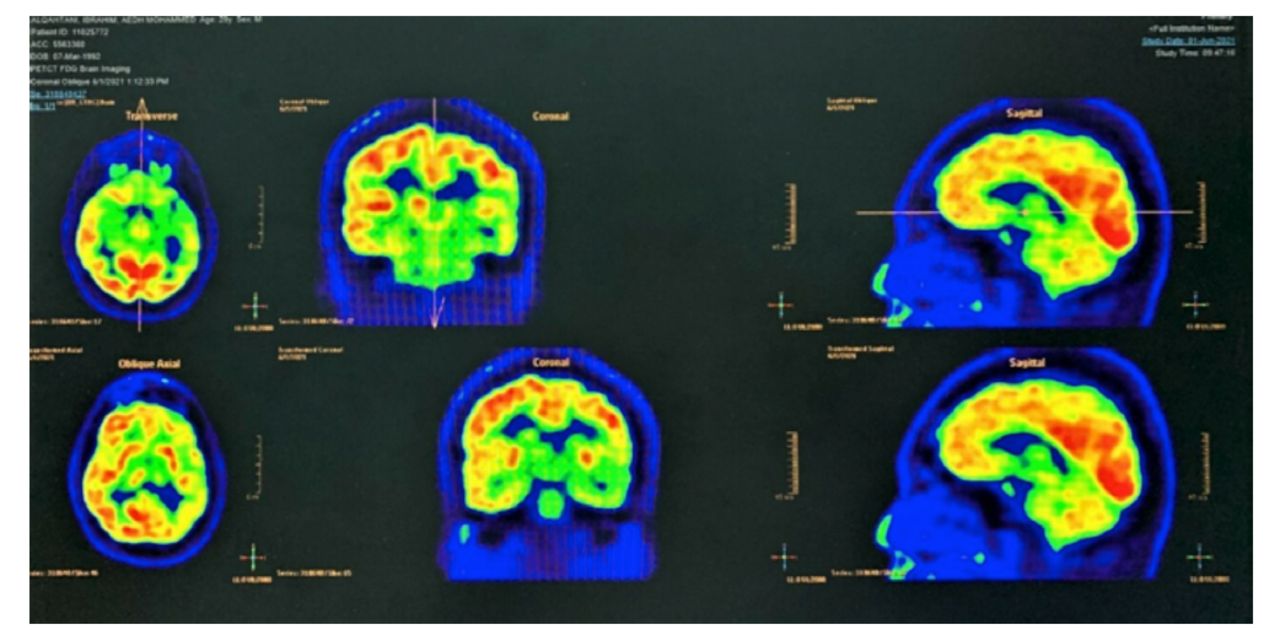
Following that, magnetic resonance imaging (MRI) of the brain was performed, which revealed left cerebral hemiatrophy with calvarial thickening. Hyperintensity in white matter represents gliosis and ipsilateral occipital horn dilatation. In addition, ipsilateral pneumosinus dilatans (frontal) is also seen. (Figure 3) Furthermore, PET/CT scans of the brain revealed diffuse hypometabolism involving the left cerebral hemisphere, more evident in the left medial temporal lobe (Figure 4).
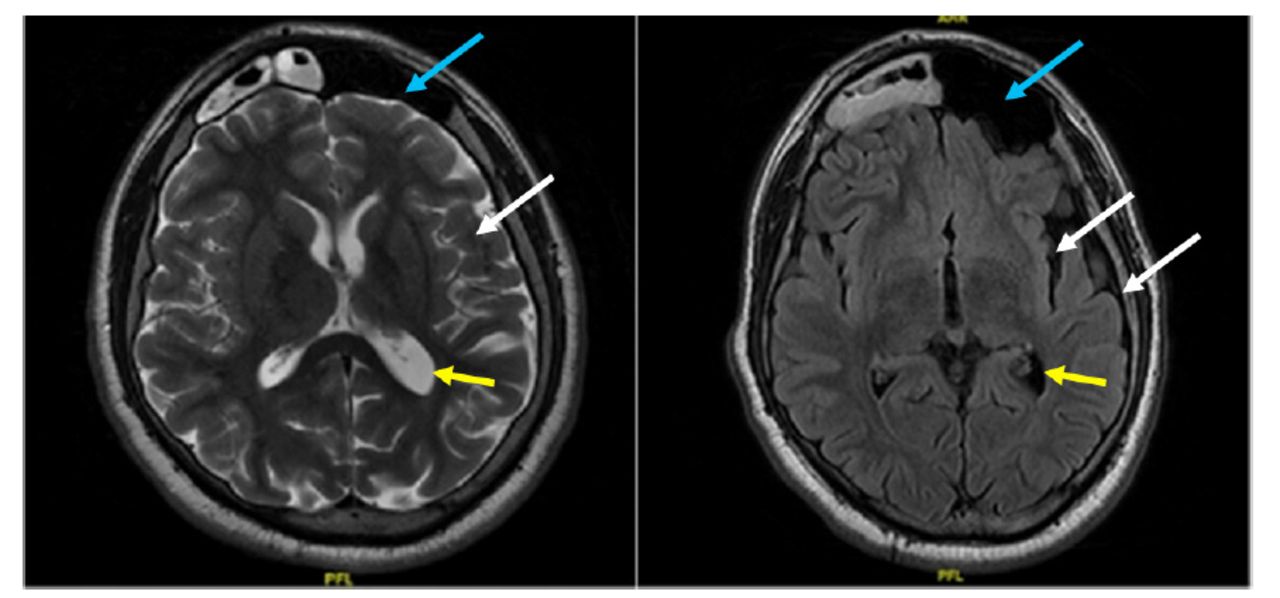
- Axial Flair/T2-weighted image: Left cerebral hemiatrophy with calvarial thickening (white arrows). Hypointensity in white matter represents gliosis and ipsilateral occipital horn dilatation (yellow arrows). In addition, ipsilateral pneumosinus dilatans (frontal) is seen (blue arrows).

- PET/CT scan of the brain revealed diffuse hypometabolism involving the left cerebral hemisphere, more evident in the left medial temporal lobe.
Therapeutic intervention, follow-up, and outcome
The patient takes carbamazepine 600 mg twice a day and levetiracetam 500 mg twice a day. During the EMU surgical discussion, the patient was found not to be a candidate for epilepsy surgery. Hence, he was advised to increase the dosage of Levetiracetam from 500 mg to 1.5 g twice daily, so the seizures were partially controlled when he was seen in the epilepsy clinic.
Discussion
The DDMS is attributed to cerebral palsy, resulting from an insult associated with immaturity of the calvarium or brain damage during childhood. Moreover, both prenatal and postnatal insults can be proposed as etiologic factors. These include cerebral infarction, hypoxia, infections, vascular malformations, congenital abnormalities, tumors, gestational vascular occlusion, especially those involving the middle cerebral vascular territory, birth trauma, intracranial hemorrhage, and prolonged febrile seizures after birth.2 The DDMS is categorized into 2 distinct types: the infantile or congenital form, which becomes symptomatic in infancy, and the acquired form. In congenital DDMS, the cerebral insult happens in utero, where midline structures move and shift to the hemisphere that contains the disease.3 Acquired DDMS occurs later in life after birth and might be caused by trauma, vascular abnormalities, hemorrhages, aortic coarctation, subependymal germinal matrix, intraventricular hemorrhage in premature infants, and amniotic bands.4
In their article, “Dyke-Davidoff-Masson syndrome: a case report,” Rashid et al5 provide a case report. In this case, a 13-year-old was suffering from episodes of focal impaired awareness seizures that were accompanied by headaches. In her case, the MRI indicated no significant value. The level of her condition was not advanced, and she did not have DRE.
Roy et al3 presented another case of “Adult Presentation of Dyke-Davidoff-Masson Syndrome: A Case Report.” The case involves a 42-year-old woman who had been born normally but later discovered to have left focal motor seizures, left-side hemiparesis, and mental retardation since the age of 2.5 years. The MRI of the brain, which was carried out on the patient, indicated atrophy of the right cerebralhemisphere, as was realized in the patient’s case. An enlarged frontal sinus and thickened calvarium were also realized on the same side. She was subjected to anti-seizure medications similar to those given to the patient in the case, but the seizures continued, which made them be attributed to poor compliance and other precipitating factors like sleep deprivation similar to those in the case.
Because of the characteristics observed after the examination, our patient has acquired DDMS. The results of MRI brain asymmetry with atrophic changes in the left hippocampus consisting of mesial temporal sclerosis indicate higher chances of the condition being acquired other than congenital. The gyral hyperintensities over the left parietal region are mainly caused by factors such as trauma or primary and secondary tumors, which are well-known causes of acquired DDMS.5 Similarly, the PET brain results indicated diffuse hypometabolism involving the left mesial temporal lobe. These results indicate reduced glucose activity in the brain, which results from neuro-generative diseases such as tumors that develop over time. However, it is challenging to determine whether the hemiatrophy emerged in childhood or came later in life.6
The MRI and PET provide the right diagnosis for both DDMS and DRE. The MRI asymmetry results assisted in the diagnosis of the condition with knowledge of its origin, whereas PET was used to determine the brain activities required to understand the condition’s risk factors.3
Rasmussen’s encephalitis, which lacks calvarial abnormalities, is the most likely alternative diagnosis based on imaging findings. Sturge-Weber syndrome is distinguished by the presence of pial angiomas and ipsilaterally dilated choroid plexuses. In hemimegalencephaly, which is characterized by a hamartomatous overgrowth of one cerebral hemisphere, the affected side has a dysmorphic lateral ventricle, whereas the contralateral ventricle in the relatively smaller and normal cerebral hemisphere is normal.7-8 The treatment of the DDMS is symptomatic, and its targets should be hemiplegia, seizures, and hemiparesis.
In conclusion, the adult presentation and treatment of DDMS are unusual and challenging. The DDMS is either congenital or acquired. However, the DDMS’s main concern remains intractable seizures, for which drug therapy is, in most cases, insufficient and a surgical approach is required. However, if the patient presents later in life, the presentation may not be similar to that seen in childhood, and management changes accordingly. Early recognition and diagnosis can help develop some remedies that would prevent the huge repercussions of the disease, mainly if it is acquired DDMS. Treatment is supportive, with the main aim of controlling seizures, along with physiotherapy, occupational therapy, and speech therapy. Moreover, psychotherapy and other alternative treatments, such as ketogenic diets and neurostimulation, may play a role.
- Received July 16, 2022.
- Accepted February 15, 2023.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.