


Abstract
Objectives: To describe the complex phenotype of ATP1A3 and second to report new mutation of ATP1A3.
Methods: This is a retrospective chart review of 7 patients who was diagnosed with ATP1A3 mutation based on whole exome sequencing (WES) result and the following information were collected; age, age of onset, developmental ability, seizure type, family history, MRI, WES report. The data collection started a year ago January 2021 in King Faisal Specialist Hospital and Research Centre, Riyadh, KSA. This has been cleared for publication by the Office of Research Affairs, and the Publication Number is 2225429.
Results: Five females and 2 males had onset ages of 0–3 years (mean=18 months). All had some degree of intellectual dysfunction, 6 had seizures (85%), 4 had neurologic abnormalities, 1 had autistic features and one had mild dystonia.
Conclusion: Our small-cohort observations confirm that ATP1A3 mutations express a wide range of phenotypes, usually including some degree of cognitive-behavioral dysfunction (100% of patients), seizures (85% of patients), and AHC (71% of patients). Moreover, they further expand the evolving allelic spectrum of these disorders by identifying 3 novel mutations.
Since the availability of exome-sequencing technology several, human genome discoveries that advance our knowledge of disease-specific pathophysiology and enable connections between genetic variants and clinical phenotypes. An example is the recent identification of variants in the ATPase Na+/K+ transporting subunit alpha 3 gene (ATP1A3), which encodes the α3 isoform for the Na+/K+ pump.
The Na+/K+ pump is heterodimeric and contains α and β subunit globular proteins. The β subunit’s main function is folding, targeting, and anchoring α into the plasma membrane, whereas the α subunit performs the main pump function. In neurons, the α3 isoform is found mostly in axons, although cell body expression has also been observed.
Variants of ATP1A3 produce a wide variety of neurological disorders. The most common symptoms are alternating hemiplegia of childhood 2012; rapid onset dystonia-Parkinsonism 1999; cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural deafness 2014; relapsing encephalopathy with cerebellar ataxia; fever-induced paroxysmal weakness and encephalopathy; and early infantile epilepsy and encephalopathy 2017. Indeed, the spectrum of disorders due to ATP1A3 variants continues to evolve, and the objective of this study is to contribute additional genotype–phenotype observations.
Methods
This is a retrospective chart review of 7 patients who was diagnosed with ATP1A3 variant based on whole exome sequencing (WES) result and the following information were collected; age, age of onset, developmental ability, seizure type, family history, MRI, WES report. The data collection started a year ago January 2021 in King Faisal Specialist Hospital and Research Centre, Riyadh, KSA. This has been cleared for publication by the Office of Research Affairs, and the Publication Number is 2225429.
Results
Five females and 2 males had onset ages of 0–3 years (mean=18 months). All had some degree of intellectual dysfunction, 6 had seizures (85%), 4 had neurologic abnormalities, 1 had autistic features, and one had dystonia. Group 1 consisted of 3 females and 2 males, with mean onset age of 24 months (Table 2). The hemiplegic episodes remitted in all of them except one (patient 5), who had the worst neurological impairment in this group, either spontaneously or after low-dose carbamazepine or flunarizine therapy. Two had mild neurologic abnormalities. Group 2 consisted of 2 females with mean onset age of 12 months (Table 3). One presented at birth with severe cognitive impairment and neurologic abnormalities, a phenotype compatible with EIEE (but no seizures), and died of apnea at 17 months old. The other presented at 2 years old with mild cognitive impairment, neurologic abnormalities, seizures, and a phenotype compatible with RECA. Notably, MRI was unremarkable in 6 patients and in one patient (patient 6) thinking of Corpus Callosum (Figure 1). Consanguinity present in 70% of parents (patient 1,2,3,6 and 7) although no positive family history of similar cases in the families, the genotype differed in all 7 patients, and 3 previously unreported de novo variant appeared (Patients 1, 3, and 5).
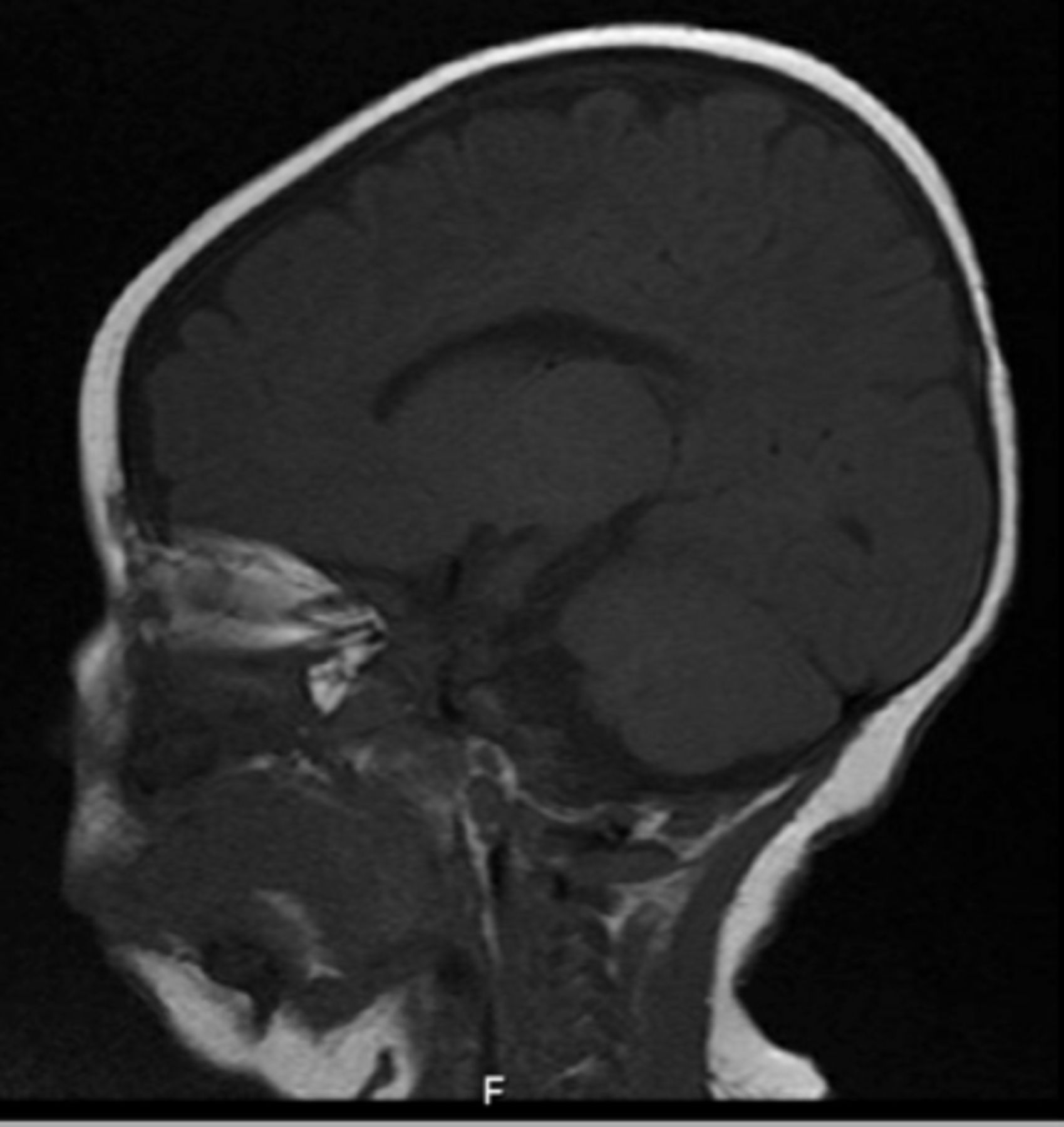
-brain MRI showing atrophy of corpus callosum (Patient 6).
- Summary of phenotypes associated with ATP1A3 gene mutation.
- Patients with AHC phenotype
- Group 2 patients without AHC phenotype.
Discussion
The clinical features in this small cohort concur with previous reports indicating that ATP1A3 variants cause a wide range of phenotypes, frequently including intellectual dysfunction, seizures, and neurologic abnormalities. Although, it is not possible to generalize from only 7 patients, the results may suggest that patients with AHC might tend to present later and have milder neurological signs than patients without AHC, and that they usually show resolution of hemiplegic episodes spontaneously or after anticonvulsant treatment and in one patient persistence of AHC seem to be associated with severe neurological dysfunction. The fact that both Group 2 patients were female while the Group 1 sex distribution was approximately equal does not necessarily indicate a gender predilection. In agreement with previous reports of genotypic variability, every patient had a different genetic abnormality, and we discovered 3 new variants.
Our small-cohort observations confirm that ATP1A3 variants express a wide range of phenotypes, usually including some degree of cognitive-behavioral dysfunction (100% of patients), seizures (85% of patients), and AHC (71% of patients). Moreover, they further expand the evolving allelic spectrum of these disorders by identifying 3 novel variants.
Acknowledgment
We would like to thank Papercheck (www.papercheck.com) for English language editing.
- Received December 21, 2022.
- Accepted June 13, 2023.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}