


Abstract
Objectives: To investigate the clinical and genetic features in a cohort of Chinese families with neurofibromatosis type 1 (NF1).
Methods: The clinical information of 21 patients with NF1 in 10 families was retrospectively analyzed. To broaden the genetic spectrum of NF1, multiplex ligation-dependent probe amplification analysis was performed first, followed by the whole-exome sequencing, in order to identify pathogenic or potentially pathogenic variants of NF1 gene in 10 unrelated Chinese families.
Results: Nine different NF1 variants were identified in all 10 families. Of these, 7 were known pathogenic variants and included the exon 1 deletion, exons 1-58 deletion, c.5401C>T (p.Q1801*), c.2291-2A>C, c.484C>T (p.Q162*), c.4922G>A (p.W1641*) and c.1019_1020del (p.S340Cfs*25). The 2 novel variants were c.5197T>C (p.S1733P) and c.783_797delinsC (p.K261Nfs*25). The p.S1733P variant was classified as a variant of uncertain significance, while p.K261Nfs*25 was classified as pathogenic. Hence, the positive detection rate of NF1 variants was 100% (10/10). While the truncating variants were responsible for 60.0% (6/10) of the cases, the splicing variant was responsible for 10% (1/10) of the cases.
Conclusion: We identified 2 novel heterozygous variants (c.5197T>C and c.783_797delinsC) in the NF1 gene, which broadens the genetic spectrum of the NF1 gene.
Neurofibromatosis type 1 (NF1, OMIM: 613113) is an autosomal dominant genetic diseases, with an estimated incidence of about 1 in 2500-5000 individuals.1 The typical clinical manifestations are skin changes including multiple café-au-lait macules (CALMs), freckling, lisch nodules (iris hamartomas), and peripheral neurofibromas.2 Previous studies have reported that the symptoms are age-dependent. While nearly half of the children with NF1 showed symptoms before the age of one, all the children were symptomatic by the time they turned 20 years.3 Furthermore, NF1 patients are prone to benign or malignant tumors, skeletal anomalies, and cognitive impairments, which result in psychological burden and nursing problems. Although NF1 is a monogenic disorder, there is high variability in the disease outcomes. The clinical symptoms can vary within a family or even at different life stages of the same patient.4-5 Therapeutic interventions include genetic counseling, surgical resection of neurofibromas and treatment with mitogen-activated protein kinase (MEK) inhibitors.5
The disease-causing gene NF1, with a 350kb genomic size and 58 exons, is located on chromosome 17q11.2 and has 3 alternatively spliced exons. NF1 is ubiquitously expressed and encodes neurofibromin, a highly conserved Ras GTPase-activating protein (GAP). It down regulates Ras/MAPK/AP-1 by converting active GTP into inactive GDP.6-7 Due to the high incidence of variants, NF1 gene has a high rate of spontaneous mutations. At least 3800 pathogenic variants have been identified till now, as per the HGMD database, (http://www.hgmd.cf.ac.uk/ac/index.php, updated on Dec 2023). The NF1 gene variants include insertions, stop and splicing mutations, whole gene deletions, amino acid changes and chromosome rearrangements. Approximately 50% of NF1 cases are due to de novo variants and lack positive family history. The de novo variants occur primarily in paternally derived chromosomes. The percentage of single nucleotide substitutions and small deletions, which include nonsense, missense or splicing mutations, was 85-90%. Moreover, large deletions or entire NF1 gene could be seen in 1-5% of the people having variants of the NF1 gene, who often had a higher incidence of cognitive impairment and dysmorphic facial features, and an earlier appearance of cutaneous neurofibromas.8 Thus, molecular testing could be helpful to identify the pathogenic variant in nearly 95 percent of NF1 patients.9 A negative genetic test might be caused by mosaicism or may represent a different disorder. Although a genotype-phenotype correlation has been observed in some NF1 cohorts, there is considerable genetic heterogeneity, inter- and intra-familial variability and dependency of phenotype on age. Further research is needed to elucidate the roles of the various causative factors in their entirety.10
In the present study, we recruited 10 unrelated Chinese patients who had been clinically diagnosed with NF1. We performed genetic investigation in these cases using multiplex ligation-dependent probe amplification (MLPA), in combination with whole-exome sequencing (WES), followed by verification using Sanger sequencing. We found 9 different NF1 variants in all 10 index patients, including 7 reported variants and 2 novel variants. The present study broadens the NF1 variants spectrum in NF1 patients from southeastern China.
Methods
From December 2020 to March 2023, 10 index patients (8 familial cases and 2 sporadic cases) with NF1 were recruited for this study. The patients’ information was collected and met the diagnostic criteria of NF1.1 Our study was approved by the Ethics Committee. Written informed consent was obtained from the participants or their legal guardians.
Genomic DNA was extracted using standard methods. Large deletions or duplications in NF1 and NF2 was examined using MLPA kits (MRC-Holland, the Netherlands), according to the manufacturer’s recommendations.11-12
The WES was performed using the methods reported previously. SIFT (https://sift.ssec.wisc.edu/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) softwares were used to predict the functional changes in proteins. The pathogenicity of the identified variants was further classified based on the criteria laid down by the American College of Medical Genetics and Genomics (ACMG). All variants were further validated by Sanger sequencing. The primer sequences for Family 2 were 5’-CTTCTCCACTTCACCCCGTC-3’ and 5’-ACTCGGGTCAGAACTGCCTA-3’; Family 4: 5’-GGCCATTCTTTACTGCACACAAA-3’ and 5’-TCCTCCTTTCTACCAATAACCGCA-3’; Family 5: 5’-CTATAGGTGTGTGCCATCATG-3’ and 5’-GACCCAGTGATTTTTTTCAG-3’; Family 6: 5’-CTATAACTGTAACTCCTGGGTC-3’ and 5’-CCCGGCTAATGTTTGTATTT-3’; Family 7: 5’-CTTAGCCTTATTTCTCAGTGTCCA-3’ and 5’-GCCTTTTGCTACCTTTGAGGC-3’; Family 8: 5’-GAAGCTGTTCAGTCTTTGTTGCT-3’ and 5’-TGCCCTCCTTACCTTATTCATGT-3’; Family 10: 5’-CTGGACAGTCTACGAAAAGCTCT-3’ and 5’-AGTAAAATCCAGCTGCCAGAAGA-3’. Cosegregation analyses were performed on all available family members.
Results
The demographic features of the 10 families, including 21 patients, are shown in Table 1 and genograms are shown in Figure 1. All the patients showed symptoms of the disorder before the age of 20; more than half of them presented skin changes before the age of 2 years. Multiple CALMs were observed in all 21 patients, and were found immediately after birth in patients 2, 7, 8, 11, 12, 17, 18, and 21. Seventeen patients suffered from cutaneous neurofibromas, and patients 4, 6, 9 and 12 had axillary or inguinal freckling. Skin lesions could be seen on the trunk in all 21 (100%) patients. Thirteen (61.9%) patients had these lesions on their limbs, while 3 (14.3%) patients had them on their necks and 5 (23.8%) on their faces. No plexiform neurofibroma was observed in any of the 21 NF1 patients. Additionally, a glioma was found in patient 3, while patients 18 and 21 had intracranial aneurysm and epilepsy, respectively. Eighty percent (8/10) of the NF1 pedigrees were consistent with autosomal dominant pattern; the 2 with a de novo variant were the only exceptions.
- Clinical data of the 21 NF1 patients in 10 families.
- Clinical data of the 21 NF1 patients in 10 families.

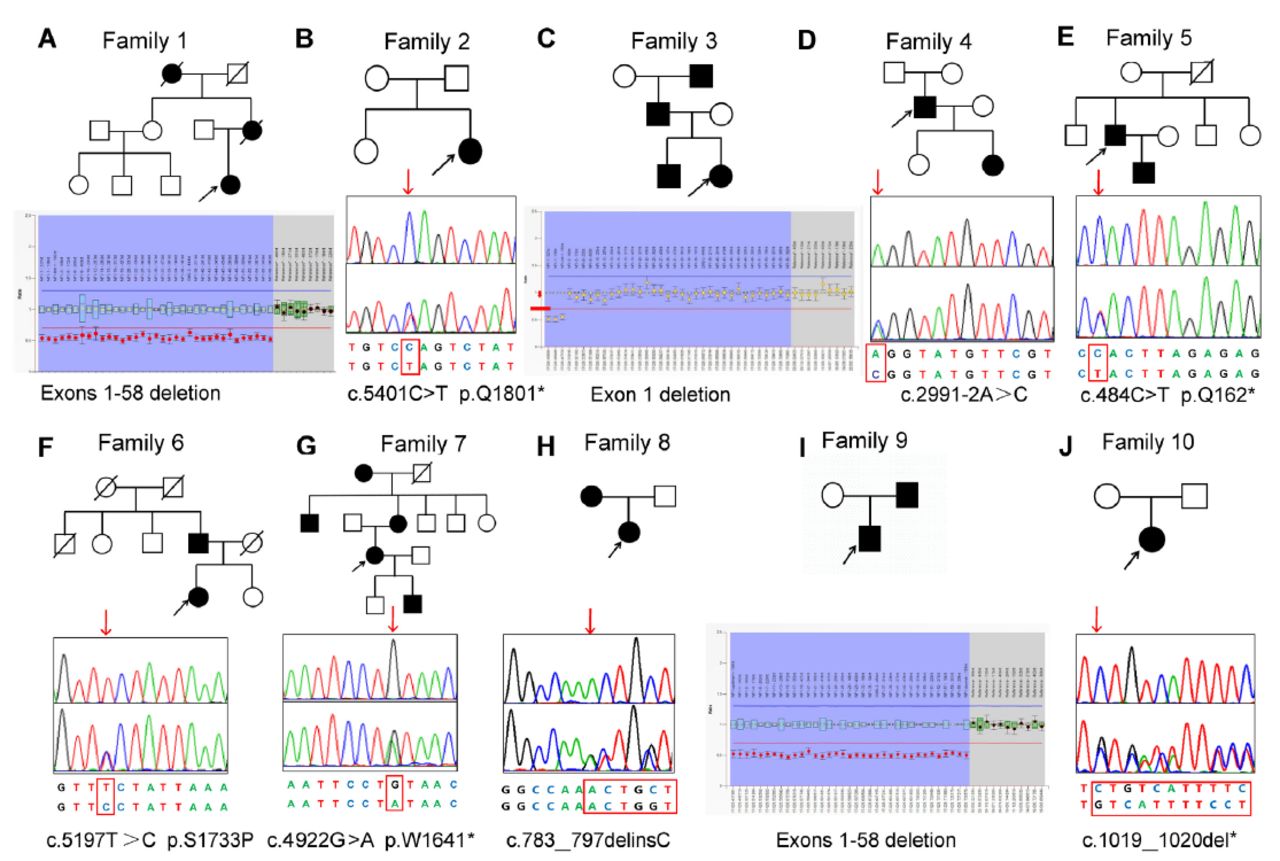
- Ten pedigree charts of families with NF1. The black symbols represent the affected individuals, and the open symbols represent the unaffected individuals. The circles and squares indicate females and males, respectively. The arrows identify the probands in the families. The second line represents the sequencing chromatograms of the NF1 variants. The upper panel in the chromatogram depicts the reference sequence. The lower panel represents the heterozygous mutated sequence.
The MLPA analysis revealed that the 3 unrelated NF1 patient probands carried an exon deletion in NF1. One patient had a single heterozygous exon 1 deletion and 2 patients had exon 1-58 deletions (Table 2). The other 7 patients with negative MLPA results proceeded to the WES analysis.
- The identified variants in the present study.
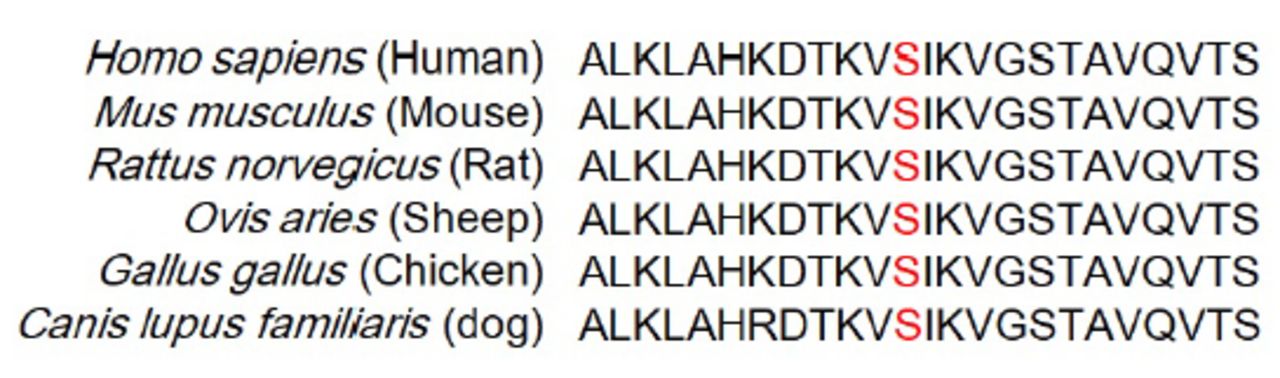
The WES was carried out in 7 NF1 probands. After filtering, 5 known pathogenic nonsense variants (NF1: c.5401C>T, p.Q1801*; c.2991-2A>C; c.484C>T, p.Q162*; c.4922G>A, p.W1641*; and c.1019_1020del, p.S340Cfs*25), and 2 novel variants (NF1: c.5197T>C, p.S1733P; c.783_797delinsC, p.K261Nfs*25) were identified in 7 NF1 patients (Table 2). All the variants were confirmed by Sanger sequencing. The novel variant was absent in GnomAD and 1000G, as well as the 500 normal controls. Based on the ACMG standards, missense variant c.5197T>C (p.S1733P) was classified as a variant of uncertain significance, while the nonsense variant c.783_797delinsC was classified as pathogenic. Multiple sequence alignment analysis showed that the residue p.Ser1733 is conserved in different species (Figure 2). All the identified pathogenic variants are summarized in Table 2.

- Sequence alignment of the p.Ser1733 residue (Red letters) of NF1 in different species.
The novel NF1 variant c.5197T>C (p.S1733P) (Figure 1F) was identified in a 25-year-old female who was born of a non-consanguineous marriage. Since birth, the girl had axillary and inguinal freckling, along with CALMs. She developed a cutaneous nodule on her back at the age of 8, with additional nodules gradually forming over time. The cutaneous nodules varied in size, ranging from 0.2 cm to 2.0 cm in diameter. She was diagnosed with NF1 after undergoing a nodule biopsy. Finally, the segregation analysis showed that her father carried this variant and also had CALMs and freckling.
The second novel NF1 variant c.783_797delC (p.K261Nfs*25) (Figure 1H) was found in a 9-year-old girl from Zhejiang province. She presented with multiple CALMs scattered on her skin at birth. Her mother found a cutaneous nodule on her trunk at the age of 5, with the number gradually increasing with age. She had a spontaneous cerebral hemorrhage when she was 9-years-old. The post-operative pathology indicated cerebral aneurysm. The co-segregation analysis indicated that her mother carried the same variant, in addition to having CALMs and freckling when she was born.
Discussion
In our current study, MLPA and WES were performed in 10 unrelated clinically diagnosed NF1 patients from southeast China. All patients were genetically diagnosed with NF1. Nine NF1 gene variants (2 of them were novel variants) were present in all 10 unrelated families. Among the variants were four nonsense variants (p.Gln1801*, p.Gln162*, p.Trp1641*, and p.S340Cfs*25), one splicing variant c.2991-2A>C, one single exon 1 deletion, one multi-exon deletion (exons 1-58 deletion), and 2 novel variants (p.S1733P and p.K261Nfs*25). The variants included nonsense, frameshift, splicing, missense and deletion variants—90% of which could result in a truncated protein and the loss of protein function. In our cohort, the detection rate of positive variants was 100% (10/10).
A truncated neurofibromin may degrade and lead to the inactivation of the protein.13 We detected 6 different truncating variants (3 nonsense, 2 frameshift, and one splicing variants), which represented 60.0% of all the variants identified. Furthermore, we detected 3 large deletion variants with a prevalence of 30.0%. The missense variant was less frequent, with a prevalence of 10.0%. Two of the detected variants (20.0%) occurred as de novo variants in the NF1 patients, which was lower than usually reported frequencies.14 Therefore, the NF1 pathogenic variants were detected in 100% (10/10) of NF1 patients, which is in line with the results of the previous studies performed in the populations from Southern Italy (96.0%, 70 of 73)15 and Spain (97.5%, 78 of 80).16 Splicing variants in NF1 account for 22-30% of NF1 patients with positive family history. Most NF1 pathogenic splicing variants cause the NF1 pathology via a haploinsufficiency mechanism, which leads to the creation of novel splice sites or the activation of cryptic splice sites within exonic and intronic sequences. This results in frameshifts and premature stop codons.17,18 Our study identified a novel NF1 splicing variant at the splice donor site of intron 22 of the NF1 gene. Furthermore, there are 6 previously reported pathogenic variants (c.2291-11T>G, c.2291-2A>G, c.2291-2A>T, c.2291-1G>A, c.2291-1G>C and c.2291-1G>T) adjacent to this region. According to the HGMD, 3872 NF1 variants have been reported worldwide to date, of which 507 are splicing variants. This suggests that splicing variants should not be ignored in NF1 patients.
A few studies from China had reported the presence of NF1 variants.5,19-23 However, no obvious correlation between the genotype and clinical features was reported in these studies.19,22 In a recent study, Zhu et al compared the clinical phenotypes with the different domains of neurofibromin in 2024. They deduced a few novel genotype-phenotype correlations in a large cohort of Chinese NF1 patients. For example, protein kinase C domain (PKC) disruption is associated with a higher rate of cutaneous neurofibromas. The patients with variations in the cysteine-serine rich domain showed a higher rate of second primary malignancy.10 However, caution should be exercised while drawing conclusions in other cohorts of NF1 patients.
Neurofibromin contains a RAS-GTPase-activating domain, which converts the active p21-RAS-GTP to the inactive p21-RAS-GDP.6 Neurofibromin regulates cell proliferation and differentiation as a RAS-MAPK signal.6,24 More than 3800 different variants have been identified in the NF1 gene until now. However, no hot-spot variants have been reported so far for this gene, with the variants occurring across the entire NF1 gene.25 Patients with NF1 variants may manifest diverse clinical characteristics even in the families harboring the same NF1 variant,26 indicating that genetic modifiers or environmental factors play an important role in the development of the clinical symptoms.27-29
In conclusion, we identified 2 novel variants and 7 reported pathogenic variants of NF1 gene in 10 NF1 families, which broadens the spectrum of NF1 variants. We acknowledge that this study has several limitations. Firstly, the sample size is small. Secondly, since this is a retrospective study, the clinical information collected may not have sufficient details to perform an exhaustive analysis. Hence, further research is required to elucidate the genotype-phenotype correlation.
Acknowledgement
We would like to thank all the patients and their families who participated in this study for their cooperation. We also thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received January 8, 2024.
- Accepted May 31, 2024.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.