


Abstract
Ataxia-Telangiectasia (A-T) is an autosomal recessive disorder caused by variants in ATM gene and characterized by progressive neurologic impairment, cerebellar ataxia, and oculo-cutaneous telangiectasia. Immunodeficiency with a recurrent sinopulmonary infections are observed in patients with A-T. Here, we report a novel stop codon variant, c.5944 C>T (p.Gln1982*), revealed by whole-exome sequencing in a 9-year old boy. He presented with recurrent upper respiratory tract infections, failure to thrive, developmental delay, ataxic gait, and bulbar telangiectasia.
Ataxia-Telangiectasia (A-T) is a multisystem disorder described in 1926 by Syllaba and Henner.1 Ataxia-Telangiectasia is characterized by a progressive cerebellar ataxia, oculo-cutaneous telangiectasia and recurrent severe sinopulmonary infections.2 Ataxia often starts in early childhood with loss of coordination as the initial manifestation of the disease. Telangiectasia, dilated small blood vessels, typically appear first in the conjunctiva then spread over the ear lobes. Immunodeficiency with recurrent sinopulmonary infection is one of the cardinal features of A-T usually observed in early childhood. Growth and endocrine abnormalities such as growth failure, delayed puberty, and diabetes have been reported in patients with A-T.3 The ATM gene causing A-T has been mapped to chromosome 11q22.3. Multiple variants in ATM gene have been identified in patients with the classical or the variant form of A-T.4 Here, we report a child with a novel variant in ATM gene who presented with typical features of A-T.
Case Report
Patient information
The proband is a 9-year old Saudi boy who attends the pediatric clinic since the age of 3 years with developmental delay and ataxia. He is a product of full-term pregnancy, born by spontaneous vertex delivery with birth weight of 2.60 kg at the 10th centile and a length of 45 cm below the third centile. His parents were first-degree cousins, and he had 2 siblings who died with undiagnosed developmental delay, unsteady gait, and cerebral atrophy. The proband was referred at the age of 8 years to our immunology clinic for further evaluation of recurrent upper respiratory tract infections. He used to get approximately 6-7 episodes a year of upper respiratory tract infections, otitis media and pharyngitis, mostly needed parenteral or oral antibiotics. His response to antibiotics was generally slow and occasionally needed a prolonged or a second course of antibiotics. He has no history of chronic diarrhea or skin infection.
Clinical findings
His developmental evaluation showed that he was 4-5 years delayed in the intellectual, speech and motor domains. His weight was 17 kg and height was 110 cm, both were below the third centile. Clinical examination revealed ocular telangiectasia and no evidence of skin lesions, lymphadenopathy or hepatosplenomegaly. Neurological examination showed intact cranial nerves, proximal muscles weakness, normal reflexes, and tone. He has impaired cerebellar signs including finger-nose test and wide ataxic gait.
Diagnostic assessment revealed normal while blood count, lymphocyte count, and peripheral blood smear. Serum immunoglobulins analysis showed a normal IgM level at 1.26 g/L (normal=0.38-2.35), IgG level at 10.2 g/L (normal=6.6-16.2) and low IgA=<0.25g/L (normal=0.57-3.18). Brain MRI showed non-specific mild atrophic changes within the cerebellar hemispheres. Table 1.
A novel variant in ATM gene causes ataxia telangiectasia revealed by whole-exome sequencing.
Molecular genetic analysis
Informed consent was obtained from the parents. The genomic DNA was isolated from the peripheral blood leukocytes of the proband and his parents. Approximately 37 Mb (214,405 exons) of the Consensus Coding Sequences (CCS) were enriched from fragmented genomic DNA by >340,000 probes designed against the human genome (Nextera Rapid Capture Exome, Illumina). The generated library sequenced on an Illumina platform to an average coverage depth 70-100X. An end to end inhouse bioinformatics pipelines in CentoGene laboratory, including base calling, primary filtering of low-quality reads and probable artifacts, and annotation of variants was applied. All disease-causing variants reported in HGMD®, in ClinVar or CentoMD® (class 1) as well as all variants with minor allele frequency (MAF) of less than 1% in ExAc database were considered. Evaluation is focused on exons and intron boundaries +/-20.
Molecular genetics result
Genomic DNA sequencing revealed a novel, homozygous variant in ATM gene. It is a nonsense substitution which interrupts the reading frame by a premature stop codon (c.5944 C>T, p.Gln1982*) (Figure 1). This variant has been detected in both parents in a heterozygous state. It is classified as likely pathogenic (class 2) according to the recommendations of the American College of Medical Genetics and Genomics (ACMG).
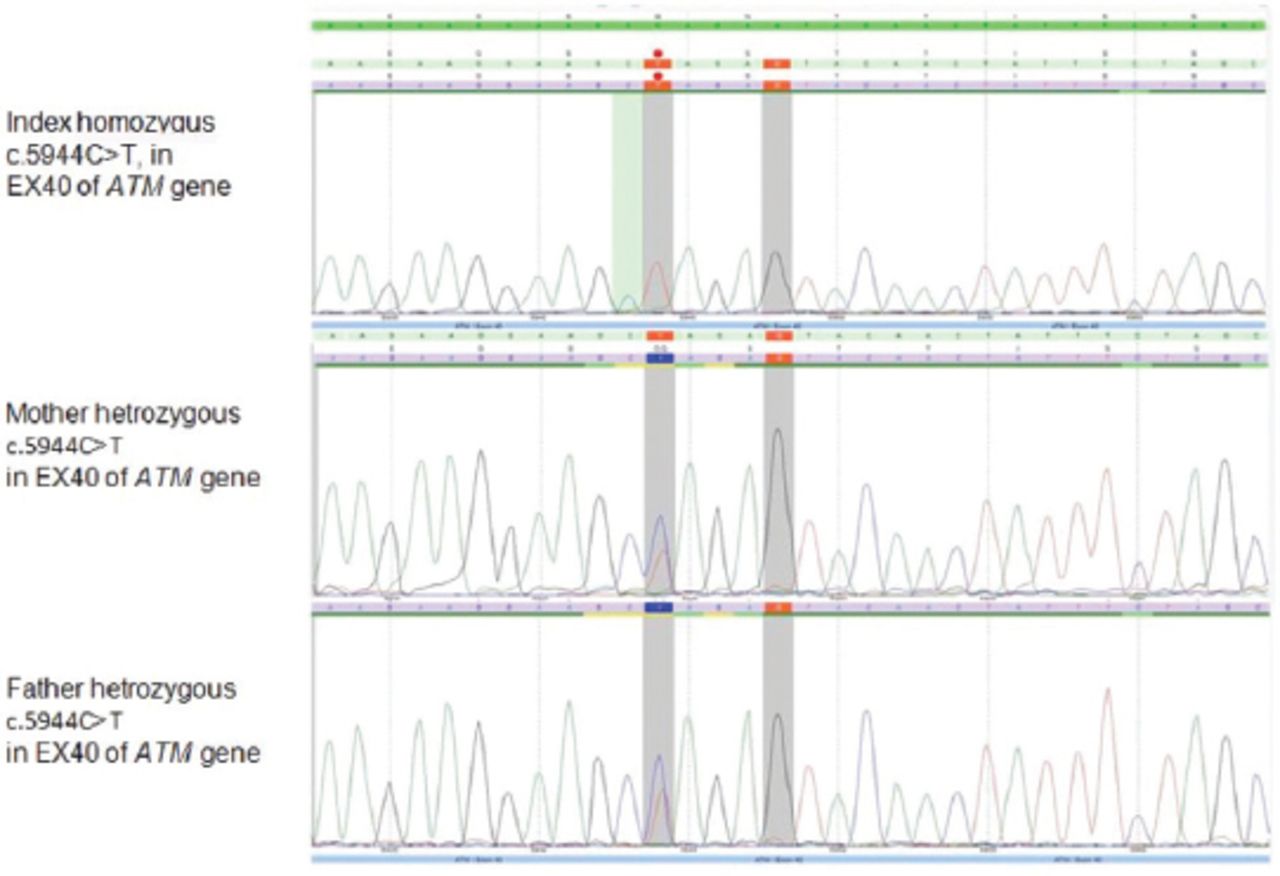
Chromatogram sequences showing homozygous variance in the index patient and heterozygous in parents.
Discussion
Ataxia telangiectasia is a rare but an important cause of ataxia in childhood. Early diagnosis of this disorder helps in tailoring and mostly avoiding complications especially those related to impaired immunity. Ataxia is the classical presentation of A-T which usually begins during toddler age group manifested as impaired coordination. Likewise, children at school age with A-T experience difficulties with reading and writing. Furthermore, patients with A-T may develop a difficulty with involuntary movement at any age, which includes chorea, myoclonic jerks dystonia and other different types of tremors.5 Our patient followed the clinical path of A-T presenting with ataxia and recurrent upper respiratory tract infections; however, it took many years to be referred to the specialized pediatric immunology clinic to confirm the diagnosis.
Telangiectasia generally becomes apparent by the age of 5-8 years, and involves the bulbar conjunctivae as in our patient. Occasionally telangiectasia can also affect other body areas which are exposed to the sun, especially the face and ears.
About 25% of patients with A-T will have pulmonary manifestations with either a cough or recurrent chest infection, or even bronchiectasis.6 Similarly, our patient had recurrent upper respiratory tract infection. Patients with A-T are at risk of developing malignancy, which occurs in 25% of all A-T patients during their lifetime. Lymphoma and leukemia are the most common types of malignancy in patients with A-T at age <16–20 years.6-8 Our patient is now under continuous surveillance regarding the risk of malignancy. Also, growth failure with short stature and poor weight gain are commonly reported in individuals with A-T as in our patient.9 The neuro-imaging studies are usually normal in early childhood years. However, as the disease progresses, the magnetic resonance imaging (MRI) studies are likely to show the pathological finding of diffuse and progressive cerebellar changes similar to those seen in our patient.10
This study reports a novel variant in ATM gene which results in premature stop codon. Unlike the previously reported variants in ATM genes which were mainly diagnosed by Sanger sequencing, we opted to use whole exome sequencing (WES) in our patient.4,6,8,10 This is because WES is readily accessible and affordable to our institute compared to Sanger sequencing.
Like our patient, Stankovic et al4 reported that 71% of patients from the British Isles with A-T had stop codon variants leading to premature termination of the protein. Compared to our patient who presented with a slowly progressive course, other patients with truncated protein tend to have a severe phenotype with an early neurological deficits.4,6,8,10 On the contrary, other types of variants as missense or splice site tend to have milder phenotype with late onset of symptoms.4,6 Despite this observation regarding phenotype/ genotype correlation in terms of type of the variants, a clinical heterogeneity among A-T patients remains the role with variable age of onset, severity of cerebellar involvement extent of immunodeficiency and predisposition to cancer.4,6,8,10 This clinical heterogeneity is illustrated by our patient who has a stop codon variant, yet he had a slowly progressive course with minimal neurological signs and apparent intact immunity and no evidence of malignancy so far.
In conclusion, this study identified a novel pathogenic stop codon variant in ATM gene in a child with the ataxia-telangiectasia. This report highlights the utility of the new generations of genetic testing in pinpointing the molecular etiology of undiagnosed genetic disorders, especially in consanguineous families that previously lost children with similar presentation.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received October 9, 2017.
- Accepted February 7, 2018.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.