


Abstract
Dysferlinopathies are rare autosomal recessive muscular dystrophies caused by mutation in the dysferlin (DYSF) gene, resulting in varied phenotype. In this case report, we review a 26-year-old diabetic male patient who presented to hospital suffering from progressive muscle weakness. We confirmed the diagnosis of dysferlinopathy with phenotype of limb girdle muscular dystrophy, followed by a muscle biopsy, immunohistochemistry and a molecular technique, exome sequencing. The specific homozygous mutations in DYSF and heterozygous mutation PSAP genes identified in the present case of LGMD-2B are found in the Saudi population. Received 10th March 2020. Accepted 25th April 2020.
Muscular dystrophies refer to a group of genetic disorders caused by dysfunction or lack of a protein in muscle cells, leading to progressive muscle weakness. The global prevalence of combined muscular dystrophies, reported in population-based studies, is one in 6200.1 Well-established muscular dystrophies include myotonic, facioscapulohumeral, limb-girdle, Emery-Dreifuss, congenital and oculopharyngeal muscular dystrophies. Dysferlinopathy is a rare autosomal recessive muscular dystrophy caused by mutation in a gene coded as dysferlin (DYSF), a plasma membrane protein found in skeletal muscles. In Saudi Arabia, the most common cause of LGMD has been reported to be mutations in the DYSF gene.2 LGMD-2B accounts for 3 to 19% of all LGMDs.3 The dysfunction of dysferlin results in necrosis of muscle fibers with the deposition of fat and fibrous tissue.4 Most often, dysferlinopathy presents in 3 of the most common phenotypes, including limb girdle muscular dystrophy type 2B (LGMD-2B), Miyoshi myopathy (MM), and distal myopathy with anterior tibial onset (DMAT).4 However, other phenotypes of myopathy associated with dysferlin deficiency have also been reported in the literature.4 Clinically, dysferlinopathy usually manifests in young adults, with slow but progressive muscle weakness and highly elevated creatine kinase.4 Although it significantly affects daily activities, it rarely involves respiratory or cardiac functions. Muscle biopsy and immunohistochemistry techniques are considered the gold standard in the diagnosis of dysferlinopathies. However, molecular techniques have improved the genetic diagnosis of such disorders. Currently, there is no cure for dysferlinopathies, including gene therapy. Therefore, families of the patients seek help to delay the progression of weakness. In this regard, supportive and immunomodulating therapy with glucocorticoids have been employed. Recently, a single case of LGMD-2B has been reported in Saudi Arabia, with clinical data to genetic analysis.5 It shows that the identification and collection of data on patients with LGMD in Saudi Arabia is lacking. This case report will be a useful addition to the literature regarding LGMD in Saudi Arabia.
Case Report
Patient information
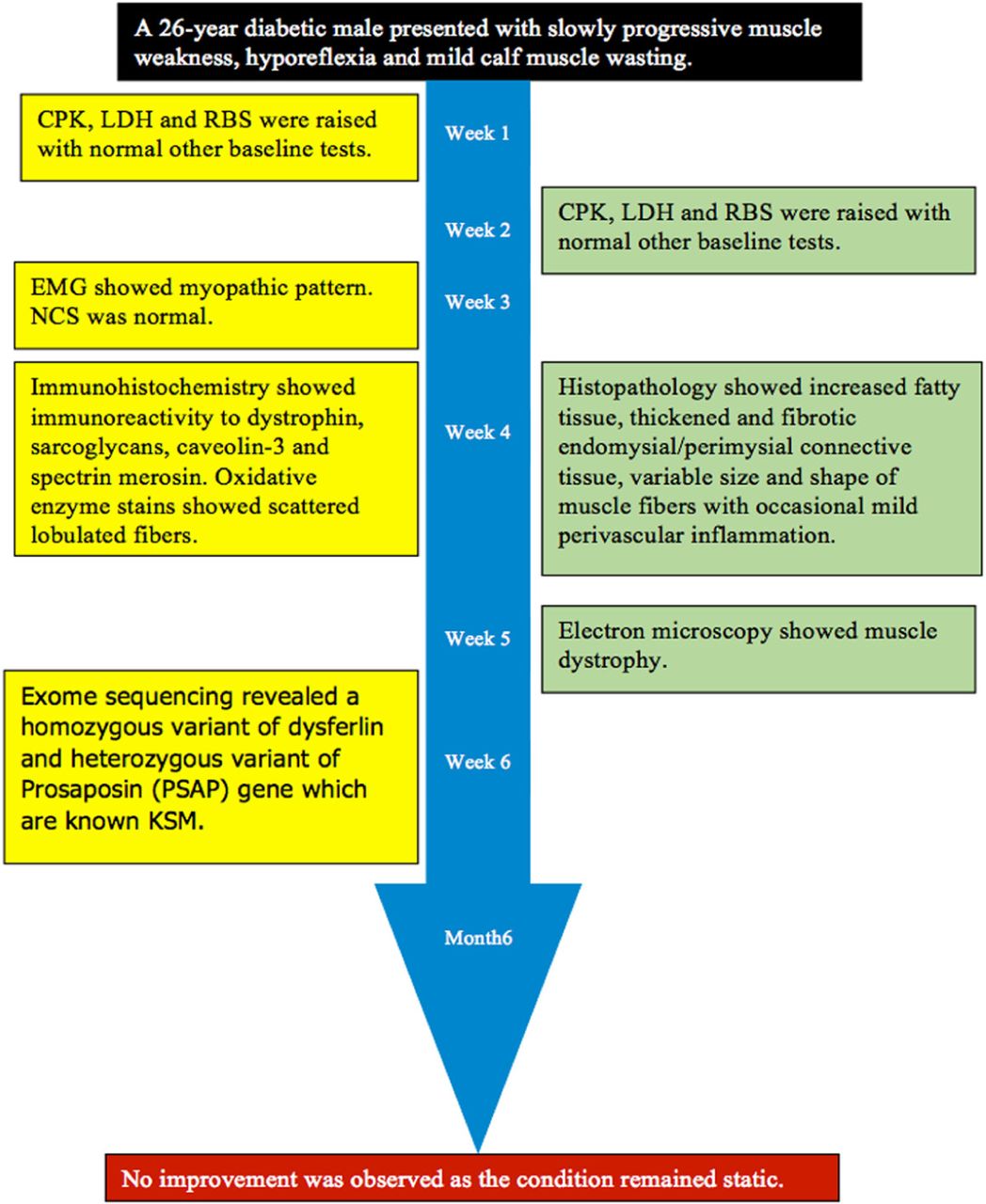
A 26-year-old male diabetic (type-1) patient presented to us in the Outdoor Patient Department (OPD) with a history of insidious onset of slowly progressive muscle weakness with diurnal variation for the last 5 years. The family history of diabetes was positive, as 2 brothers of the patient were suffering from diabetes. His medical and psychosocial history was not significant. The father of the patient was deceased and the patient’s mother refused to undergo genetic testing.
Clinical findings
On examination, the patient was alert, and his vitals were stable. There was global weakness of upper and lower primary muscles, where the muscle weakness was more proximal than distal, with hyporeflexia, hypotonia and mild calf muscle wasting (atrophy). Muscle strength grade of flexors and extensors of fingers, wrist and elbow was 4/5 bilaterally, while the muscle strength of shoulder adductors and abductors was at 3/5 bilaterally. Similarly, the muscle strength grade of ankle plantar flexors, ankle dosiflexors, knee extensors and flexors was 4/5 bilaterally, while the muscles for hip adduction, abduction, flexion and extension had a muscle strength grade of 3/5 bilaterally. The patient had a normal sensory examination with no ocular or swallowing problems. There was no clinical evidence of myasthenia gravis.
Diagnostic assessment
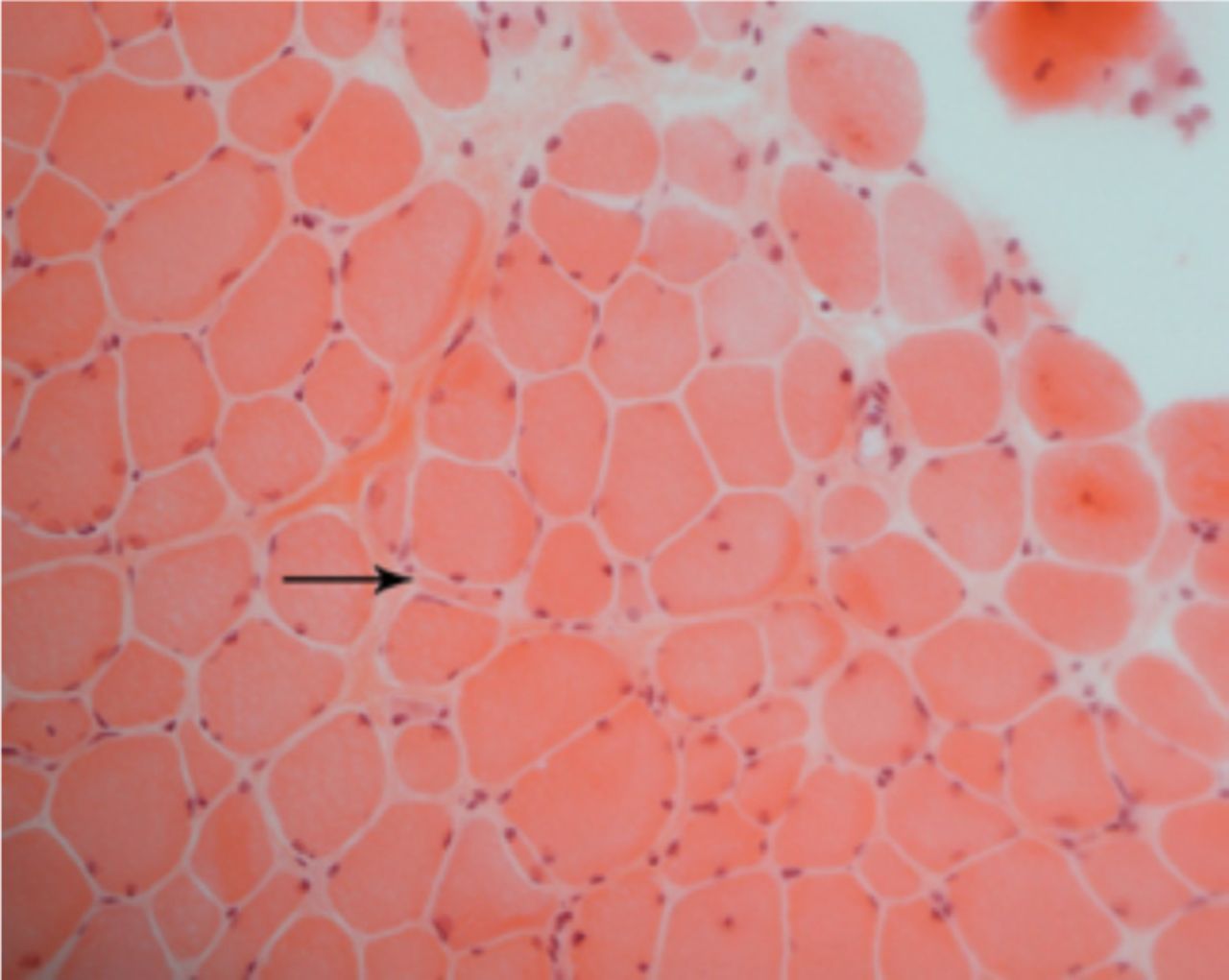
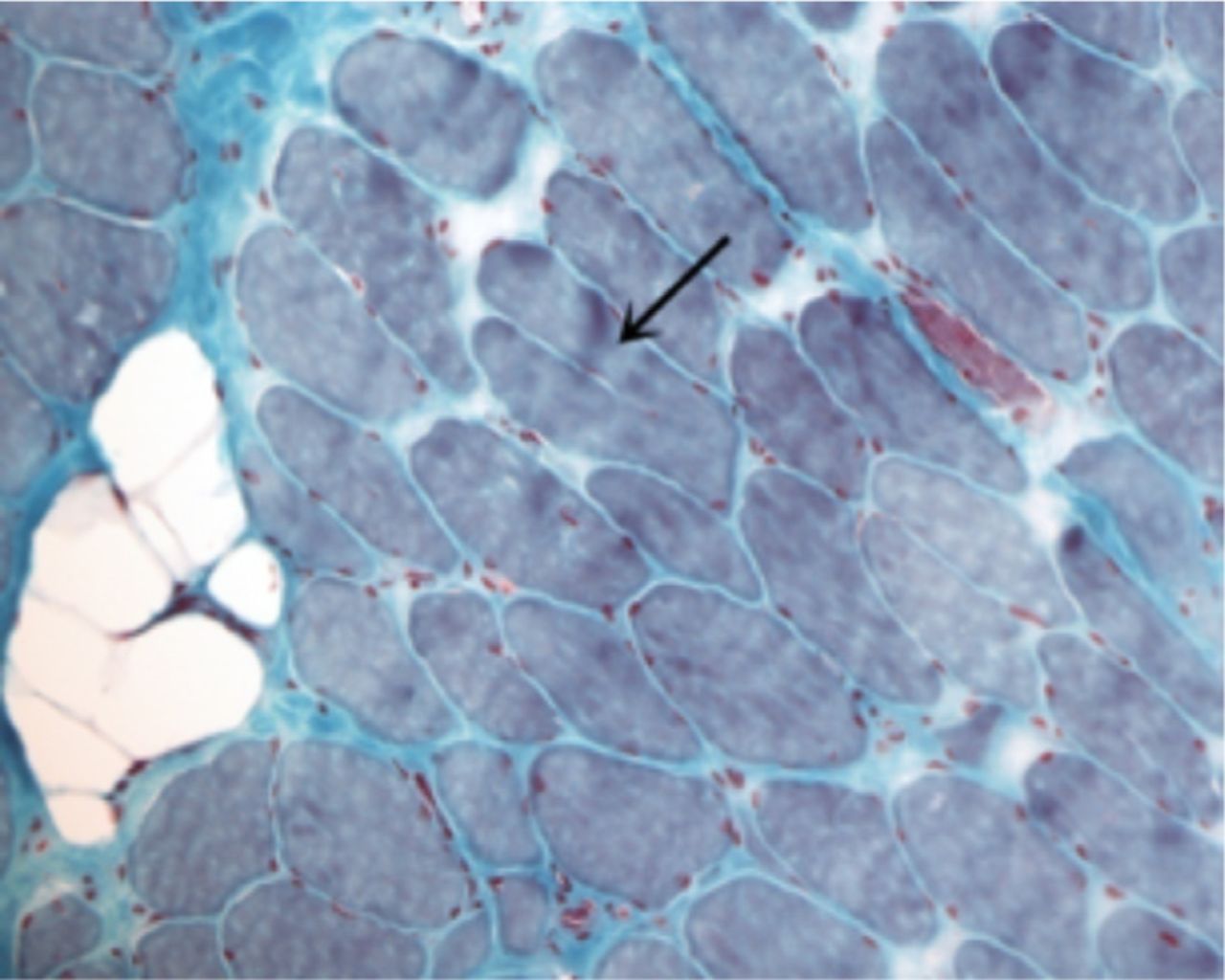
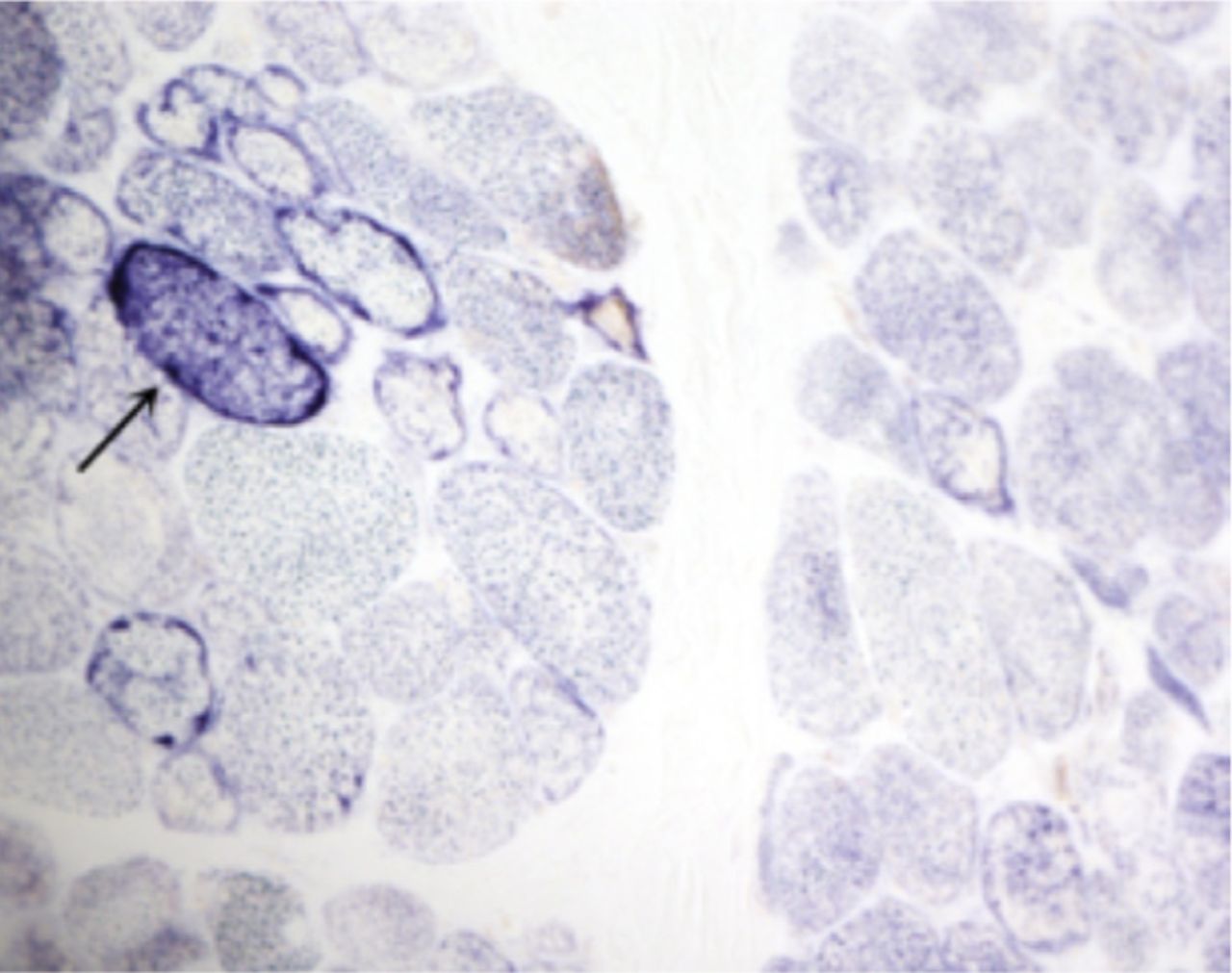
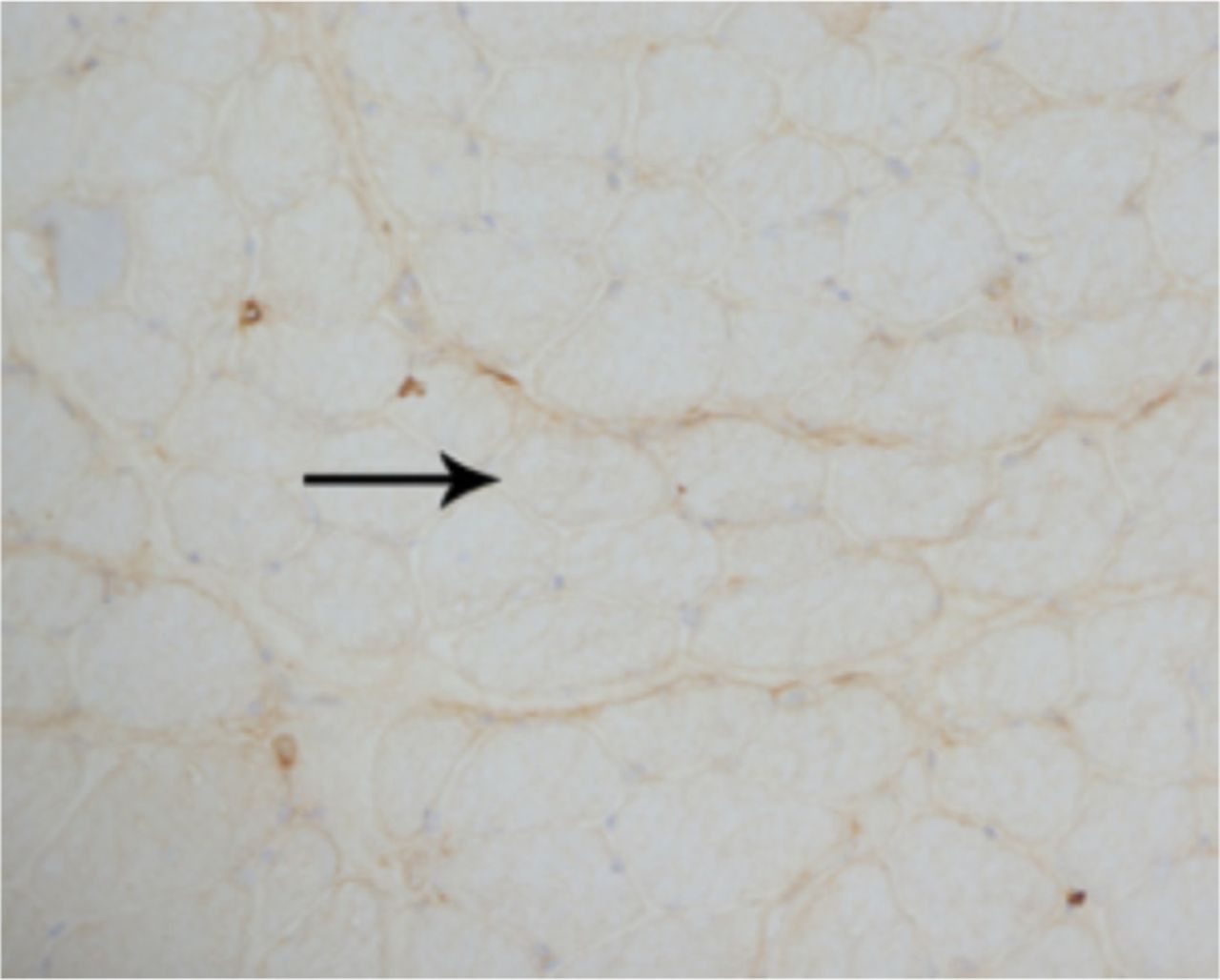
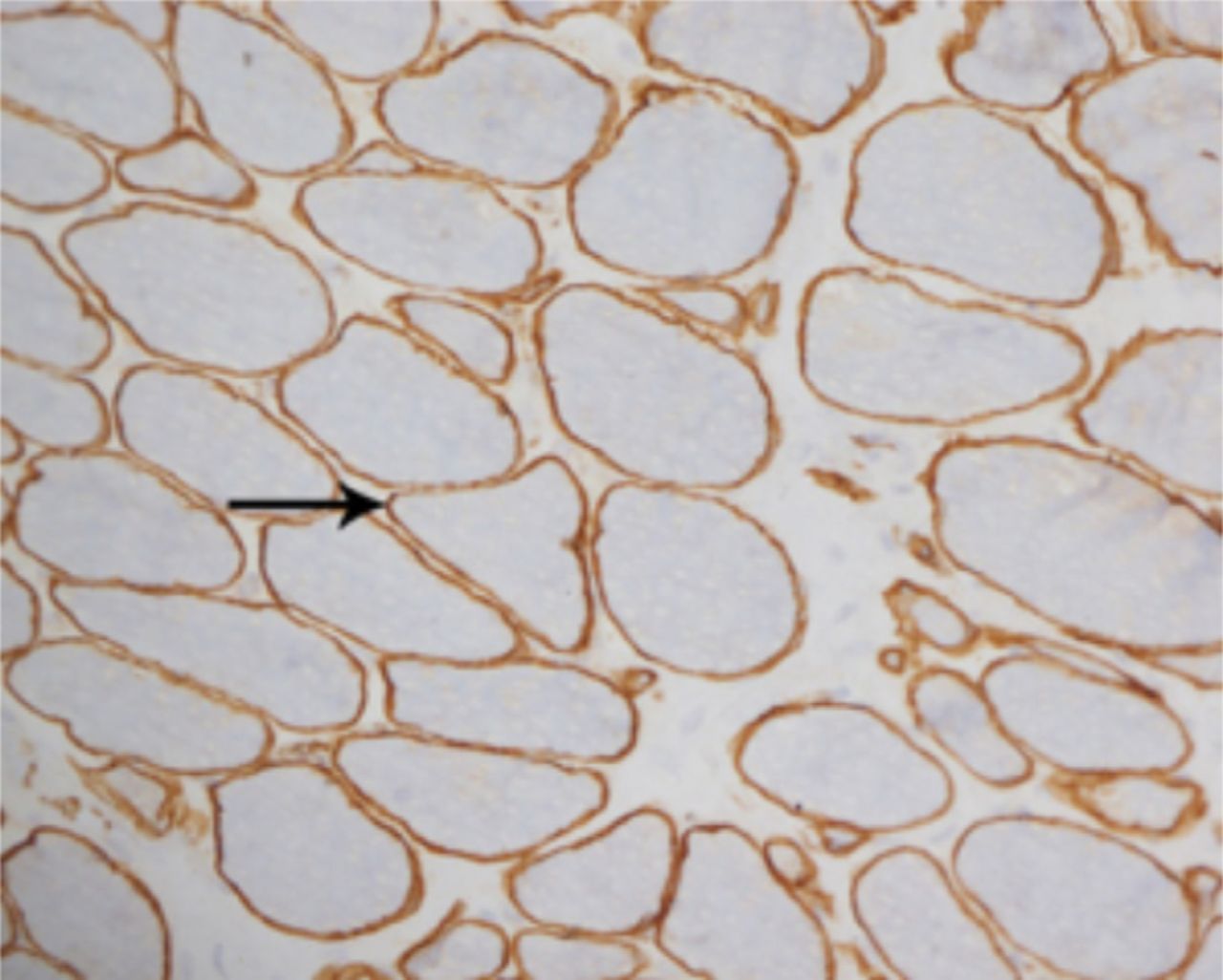
A laboratory investigation found that his creatine kinase (CK) and lactate dehydorgenase (LDH) levels were 14110 U/L and 5121 U/L, respectively. His fasting blood sugar was 10.92 mmol/L, and his acetylcholine receptor (AChR) antibody was borderline. Electromyography (EMG) showed a myopathic pattern, while a nerve conduction study (NCS) turned out to be normal. His complete blood count (CBC), renal parameters (RPMs), erythrocyte sedimentation rate (ESR), vitamin B12, vitamin D and thyroid function tests (TFTs) were in normal ranges. Histopathology of skeletal muscle revealed a mildly increased fatty tissue component and moderately thickened and fibrotic endomysial and perimysial connective tissue, with occasional mild perivascular inflammation (Figures 1 & 2). The muscle fibers showed variability in shape and size, ranging from small to hypertrophic. Oxidative enzyme stains (e.g. NADH) showed scattered lobulated fibers (Figure 3). Overall, the muscle presented a dystrophic appearance. On immunohistochemistry analysis, all fibers showed strong immunoreactivity to dystrophin, sarcoglycans, caveolin-3, spectrin (Figure 5) and merosin. Emerin showed strong nuclear positivity, while dysferlin immunostain was negative (Figures 4). Electron microscopy showed features in keeping with a dystrophy. Exome sequencing revealed a homozygous variant of the dysferlin (DYSF) gene (DYSF: NM_001130455:exon3:c.166dupA:p.L55fs) and a heterozygous variant of Prosaposin (PSAP) gene (NM_001042465:exon7:c.722G>C:p.C2415). Both of these mutations are known Saudi mutations (KSMs), which are pathogenic in nature.
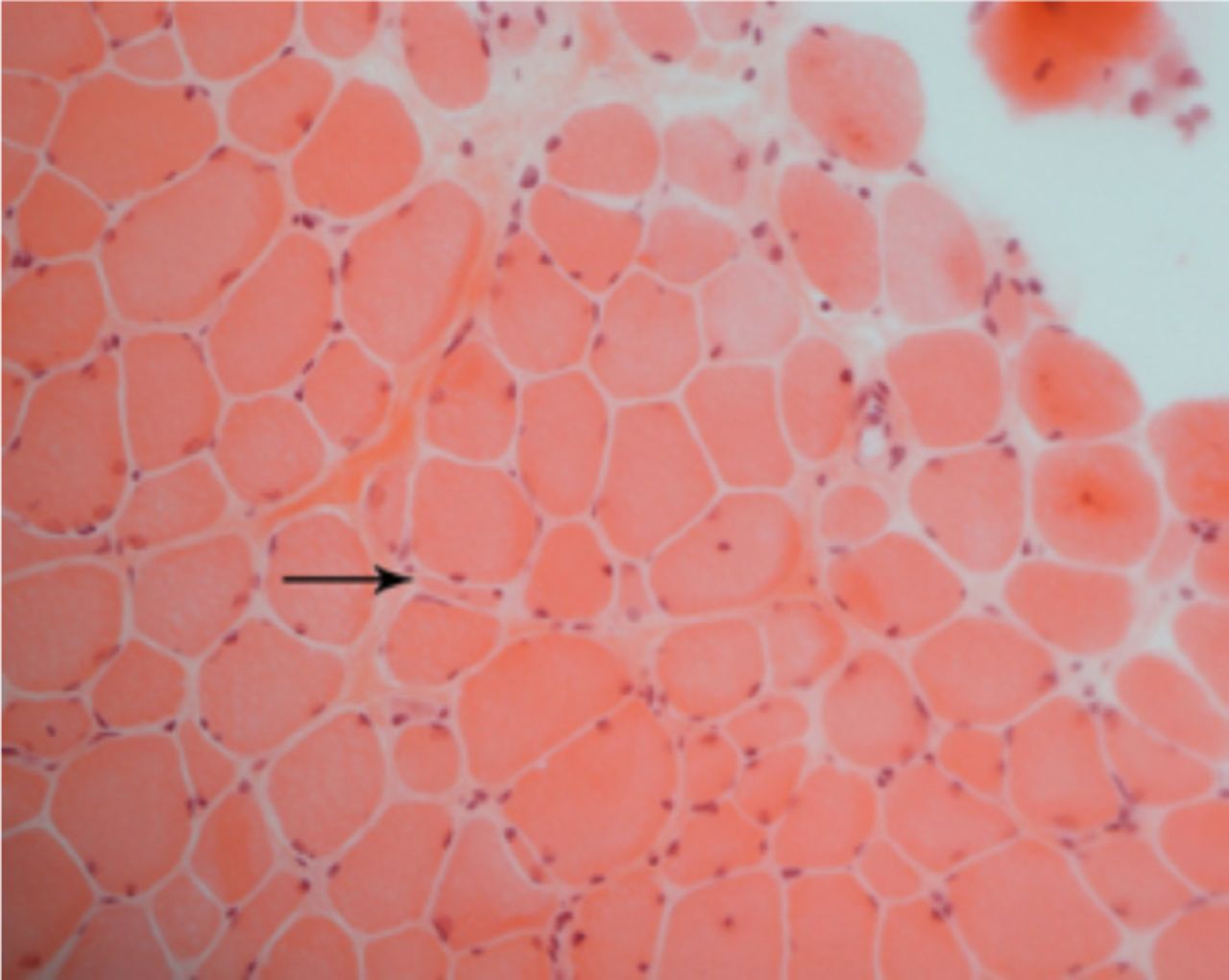
Light microscopy shows marked variation of fiber size, with scattered atrophic and hypertrophic fibers and increased endomysial fibrosis (H&E, X200)

Trichrome stains highlight increased endomysial fibrosis, with a focal fat component. Split fiber is present in this field. (arrow, Trichrome, X200)
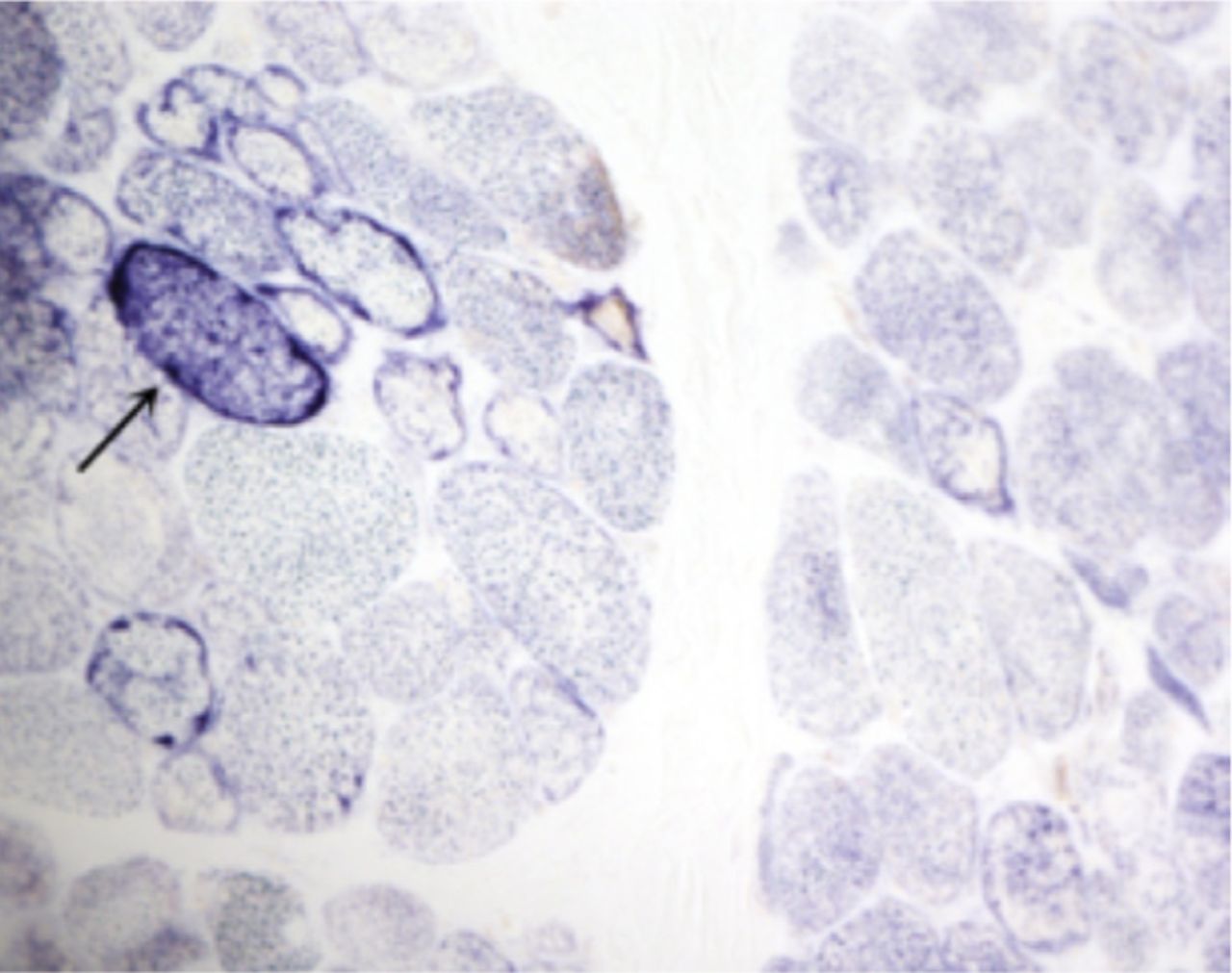
An oxidative enzyme stain (NADH) revealing scattered lobulated fibers (arrow, NADH, X200)
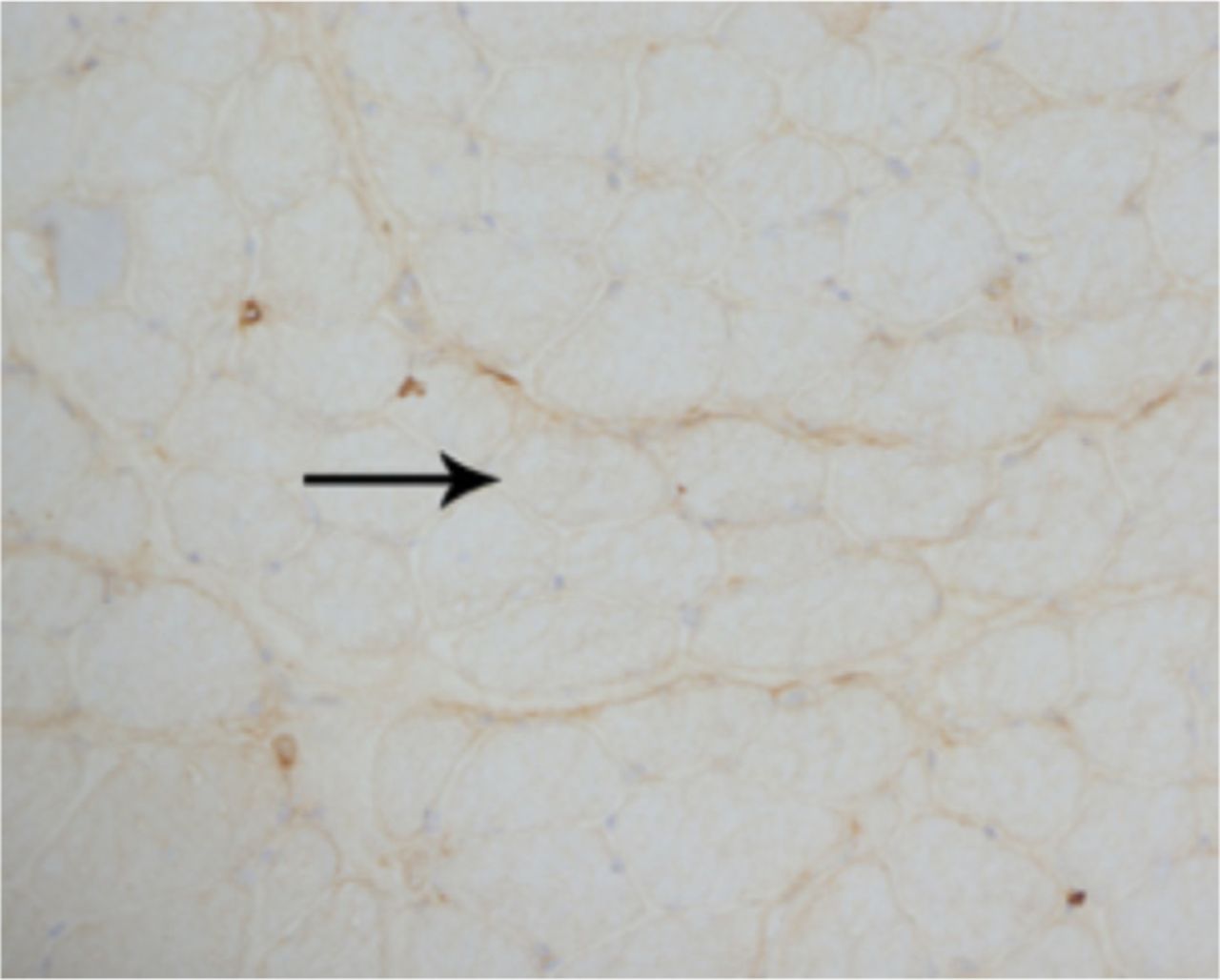
The fibers show almost complete loss of membranous reactivity to dysferlin immunohistochemical staining (X200)
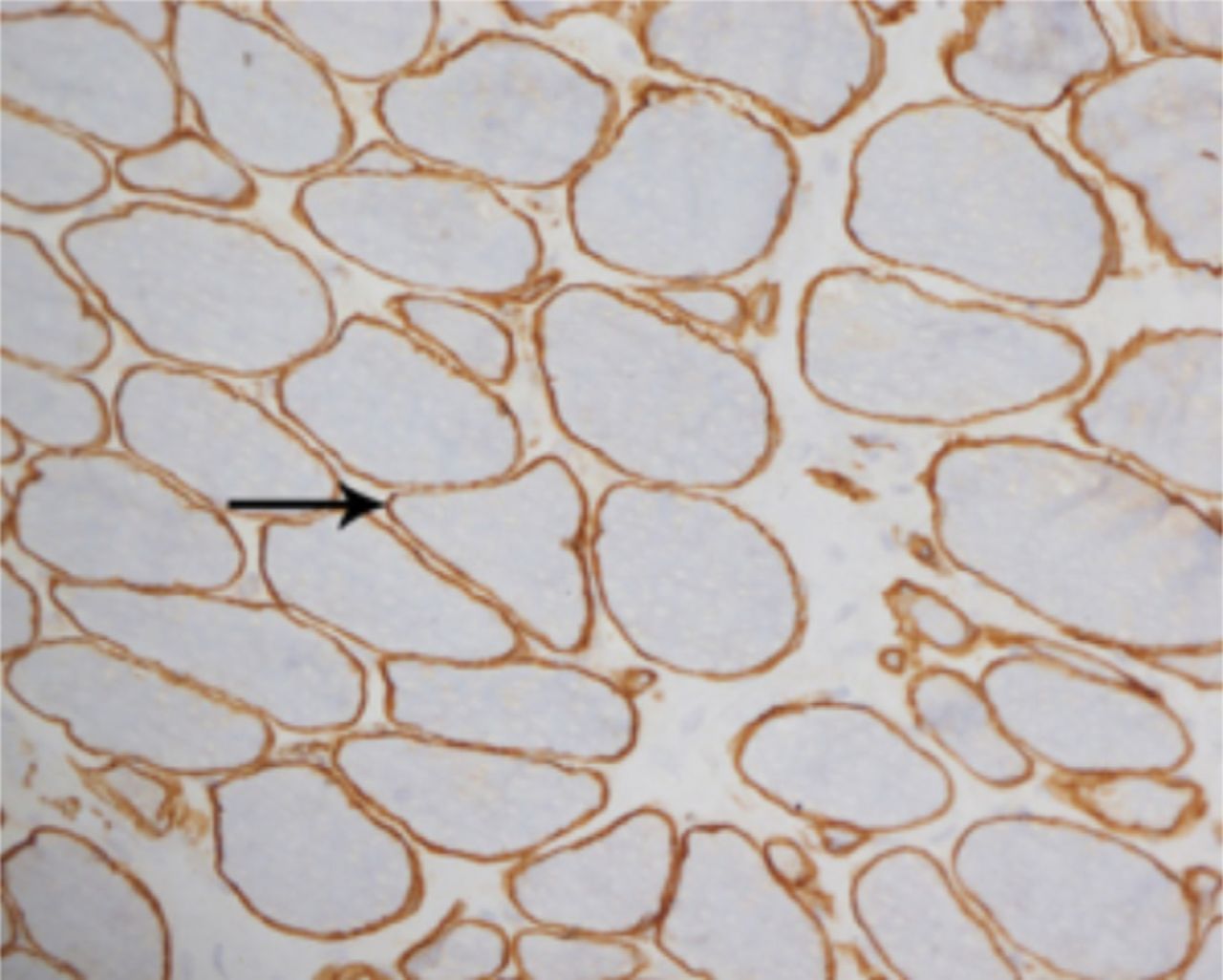
For comparison, the muscle fibers show normal membranous reactivity to spectrin (X200)
Therapeutic intervention
To date, there is no known treatment for LGMD-2B. However, thorough counseling was offered to the patient regarding optimal management of diabetes. The patient was referred to a disability social worker for supportive management.
Follow up and outcomes
At the 6-month follow-up, no improvement was observed, as the condition had remained static. However, CK was 11713 U/L (Figure 6).
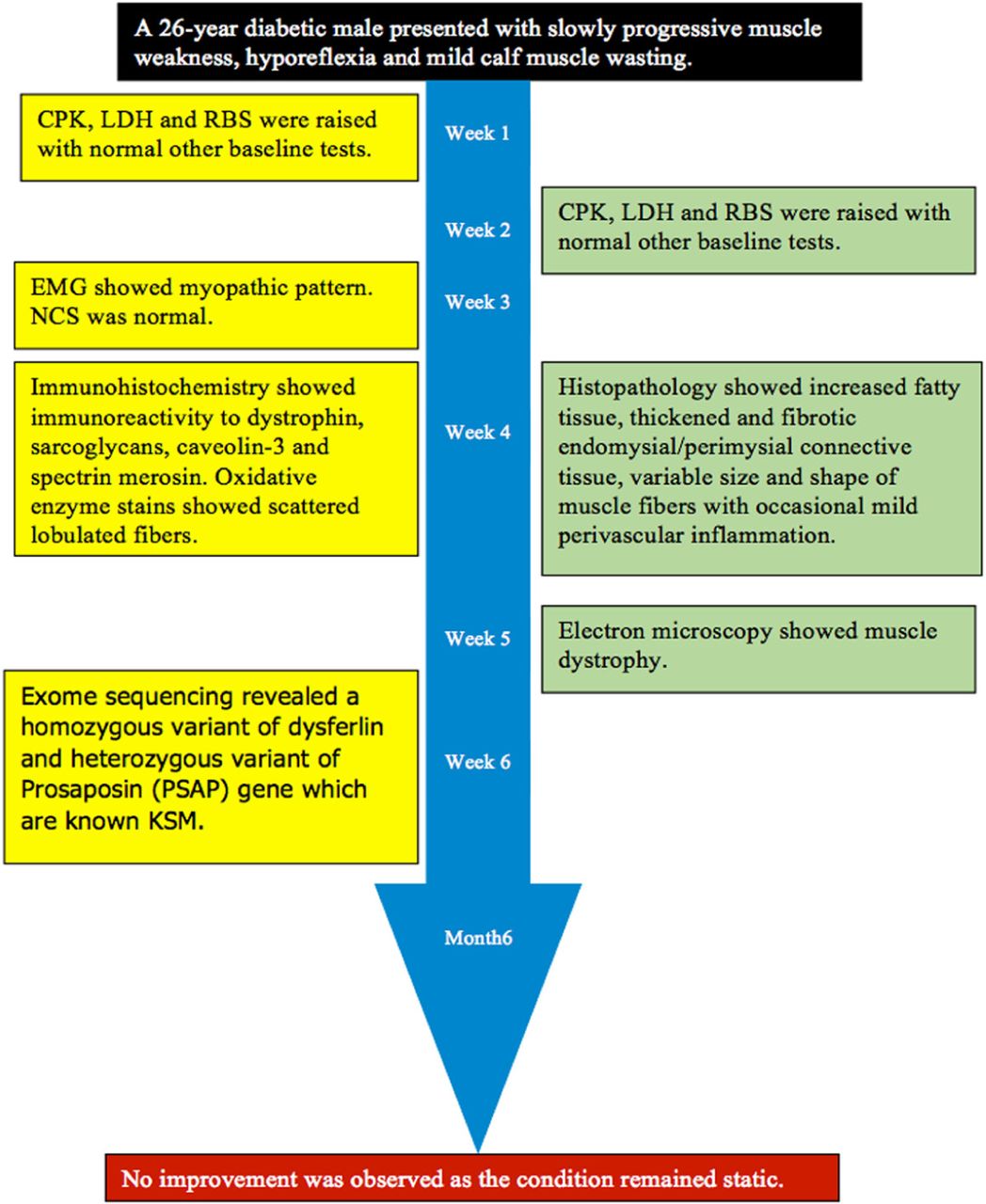
Timeline of a 26-year-old male diabetic (type-1) patient presented to us in the Outdoor Patient Department (OPD) with a history of insidious onset of slowly progressive muscle weakness with diurnal variation for the last 5 years.
Discussion
Dysferlinopathy is a spectrum of muscle diseases with several different clinical presentations. The specific homozygous mutations in DYSF and heterozygous mutation PSAP genes identified in the present case of LGMD-2B are found in the Saudi population. So, these are included in the list of known Saudi mutations (KSMs).6 The DYSF gene encodes dysferlin, a protein involved with skeletal muscle repair, while the PSAP gene encodes prosaposin, a protein involved in the development of the reproductive and nervous systems.4,7 The importance of these mutations is that these are pathogenic and present with clinical manifestations. The patient had a homozygous mutation in DYSF, and developed a form of proximal muscular dystrophy.
Recently in Saudi Arabia, Algahtani et al5 reported an LGMD-2B case where a 41-year-old woman presented with insidious onset of proximal muscular weakness. Genetic analysis showed a duplication in the DYSF gene. In the United States, Patel et al.8 reported a 21-year-old female with LGMD-2B and a 25-year-old male with Miyoshi myopathy. In addition, LGMD-2B may be mistaken as polymyositis. Xu et al.9 reported a 25-year-old female with LGMD-2B, who was first diagnosed with polymyositis. They demonstrated that a combined application of immunohistochemistry and Western-blot analysis assists the early detection of LGMD-2B. Literature on the frequency of LGDM-2B and other muscular dystrophies within the population of Saudi Arabia has not been published. In Israel and Algeria, LGDM-2B makes up 20% and 13.1% of all muscular dystrophies, respectively.10
In addition, the individual was a carrier of the PSAP variant (exon 7: c722G>C:pC2415), and he was heterozygous to this variant. Being a heterozygous condition, it would cause a disorder associated with mutations in PSAP. The defective PSAP protein is associated with autosomal recessive conditions such as prosaposin deficiency, saposin B deficiency, Gaucher disease, Krabbe disease, and metachromatic leukodystrophy.11⇓–13 Therefore, the patient should also be checked for these disorders, along with LGMD-2B. Moreover, the present case report indicates that dysferlinopathies may be associated with other genetic disorders.
Acknowledgements
This publication was supported by the Deanship of Scientific Research at Prince Sattam bin Abdulaziz University, Alkharj, Saudi Arabia
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company. This publication was supported by the Deanship of Scientific Research at Prince Sattam bin Abdulaziz University, Al-Kharj, Kingdom of Saudi Arabia
- Received March 10, 2020.
- Accepted April 25, 2020.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.