


Abstract
Hashimoto’s encephalopathy is a rare neurological syndrome occurring in patients with autoimmune thyroid disease. The diagnosis of Hashimoto’s encephalopathy is based on the clinical picture with the presence of serum anti-thyroid antibodies regardless of the thyroid disorder. Acquired cerebellar ataxia associated with Hashimoto’s disease is a rare occurrence. In this article, we present a case who had progressive non-familial autoimmune pancerebellar disease in association with an increased level of thyroid peroxidase and thyroglobulin antibodies. The patient was managed aggressively with both intravenous immunoglobulins and plasma exchange, which stopped the progression of the disease and allowed for slow improvement. Early diagnosis of Hashimoto’s encephalopathy with autoimmune cerebellar ataxia and intervention with immunomodulatory therapy are of paramount importance. Close monitoring after steroid therapy is important since some patients with this rare disease might be resistant to steroid therapy and require aggressive immunomodulatory therapy.
Hashimoto’s encephalopathy is a rare neurological syndrome occurring in patients with autoimmune thyroid disease.1 The diagnosis of Hashimoto’s encephalopathy is based on the clinical picture with the presence of serum anti-thyroid antibodies regardless of the thyroid disorder.2 Acquired cerebellar ataxia associated with Hashimoto’s disease is a rare occurrence.3 In this article, we present a case who had progressive non-familial autoimmune pancerebellar disease in association with an increased level of thyroid peroxidase and thyroglobulin antibodies. The patient was managed aggressively with both intravenous immunoglobulins and plasma exchange, which stopped the progression of the disease and allowed for slow improvement.
Case Report
Patient Information
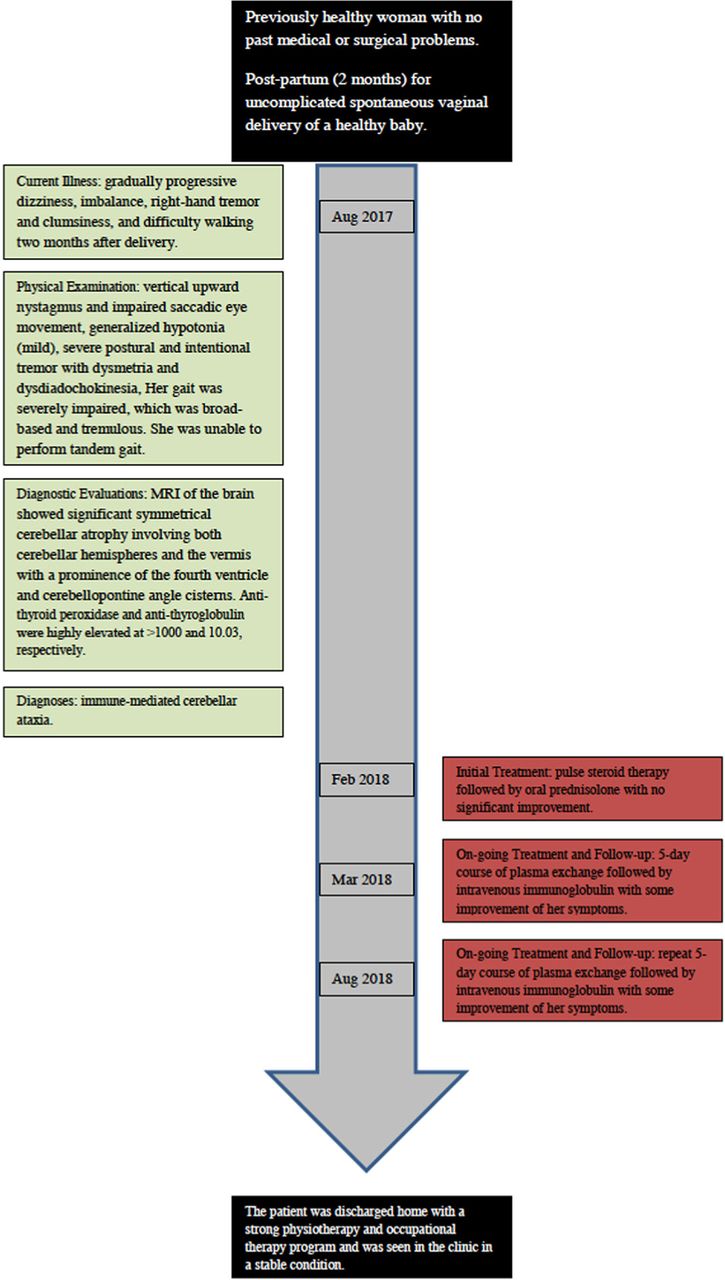
A 25-year-old female presented to the neurology clinic with gradually progressive dizziness, imbalance, right-hand tremor and clumsiness, and difficulty walking 2 months after delivery. She delivered a healthy girl through uncomplicated spontaneous vaginal delivery. She is a mother of a 3-year-old boy with a previous history of one abortion and ABO incompatibility in her first pregnancy. There was no history of seizures, forgetfulness, sensory symptoms, weakness, or bowel/bladder involvement. Her family history was unremarkable for any neurological diseases including hereditary ataxias. Her symptoms were progressive with difficulty mobilizing without support, and she required a wheelchair 6 months after the onset of her disease. Her speech was slurred and her tremor caused difficulty in feeding and other activities of daily living. She also developed severe visual impairment due to oscillopsia.
Clinical Findings
Her neurological examination showed normal higher mental functions. Her cranial nerve examination showed vertical upward nystagmus and impaired saccadic eye movement with normal facial motor and sensory functions. Her visual acuity, pupils size and reactivity, and hearing were all normal. Her motor examination showed generalized hypotonia (mild) with normal power and reflexes. Her coordination examination showed severe postural and intentional tremor with dysmetria and dysdiadochokinesia. Her gait was severely impaired, which was broad-based and tremulous. She was unable to perform tandem gait. Scale for the assessment and rating of ataxia (SARA) was used, and she scored 33/40, which indicates severe ataxia. Her cerebellar findings were symmetrical bilaterally. All sensory modalities were normal.
Diagnostic Assessment
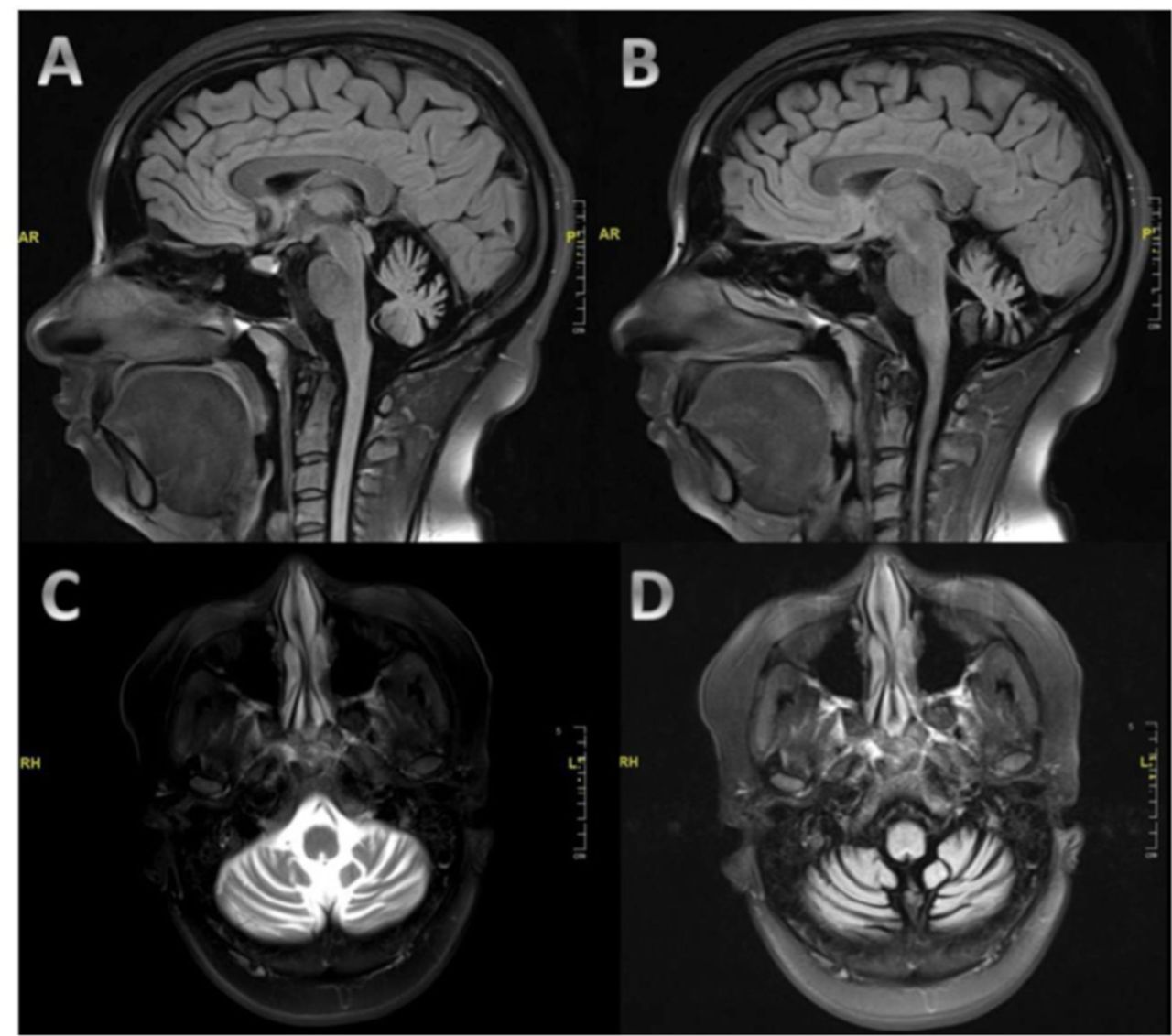
MRI of the brain showed significant symmetrical cerebellar atrophy involving both cerebellar hemispheres and the vermis with a prominence of the fourth ventricle and cerebellopontine angle cisterns (Figure 1). The cerebral hemisphere, basal ganglia, and brainstem structures were normal. Systemic investigations, connective tissue screen, celiac disease autoantibodies, and thyroid function tests were all normal. Workup for Wilson disease and organic acid disorders were negative. Anti-thyroid peroxidase and anti-thyroglobulin were highly elevated at >1000 and 10.03, respectively, which is consistent with the diagnosis of Hashimoto’s disease. Serial investigations for malignancy and paraneoplastic syndrome were unremarkable including computed tomography scan of the chest, abdomen, and pelvis, tumor markers, and paraneoplastic serological tests (e. g. anti-Ri, anti-Hu, anti-Yo). Serum protein electrophoresis did not show an increase in IgM and no monoclonal spikes. Genetic testing panel for hereditary ataxia was negative. Investigations for connective tissue disease was negative including anti-nuclear antibody, rheumatoid factor, and antibodies for Sjogren’s syndrome (anti-SS-A and anti-SS-B). Investigations for other immune-mediated cerebellar ataxia including anti-GAD, anti-NMDA receptor antibodies, anti-Purkinje, and anti-neuronal nuclear were negative. Cerebrospinal fluid (CSF) assessment was unremarkable with negative oligoclonal bands. The CSF was not tested for anti-thyroid peroxidase and anti-thyroglobulin antibodies.

MRI of the brain demonstrating moderate to severe cerebellar atrophy. The images are sagittal T1-weighted images (A and B), axial T2-weighted images (C), and axial T1-weighted images (D).
During the admission, she was found to have thrombocytopenia, at which point the valproic acid was discontinued. On the day after valproic acid was discontinued, the spasms stopped, although ACTH was not given. Repeat EEG 1 week later showed focal slowing over the right side with the resolution of the hypsarrhthmic pattern (Figure 2).

Timeline figure for our case.
Therapeutic Intervention
The patient was treated with pulse steroid therapy followed by oral prednisolone with no significant improvement. She was then treated with a 5-day course of plasma exchange followed by intravenous immunoglobulin with some improvement of her symptoms and SARA scale assessment (25/40) for 6 months. She was also treated with 4-aminopyridine for one month with no improvement in her condition. 4-aminopyridine was discontinued due to lack of efficacy according to the patient.
Follow-up and Outcomes
Unfortunately, we had to repeat the immunomodulatory therapy due to relapse of her symptoms, but again with some improvement. The patient was discharged home with a strong physiotherapy and occupational therapy program and was seen in the clinic in a stable condition (Figure 2).
Discussion
Disorders of the thyroid gland accompanied by neurological complications are common with a frequency of 1-2% of unselected general medical, geriatric, and psychiatric in-patients.4 Neurologists should be aware of the common, rare, and unusual neurological complications of thyroid disease. This is because neurological complications may be the presenting features of thyroid disorders and they are usually readily treatable with appropriate early therapy. The neurological complications of thyroid disease may result from an immune-mediated mechanism or from an alteration in the levels of circulating thyroid hormones. Hormonal alterations may exacerbate a pre-existing neurological disorder, increase the severity of a subclinical problem, or produce a new neurological disorder.5 Other mechanisms resulting in neurological complications include local compression of adjacent structures from enlargement of the thyroid gland, development of cerebral metastasis from thyroid carcinoma, and raised intracranial pressure in patients with hypothyroidism.6
The first description of the possible association between Hashimoto’s thyroiditis and the development of an autoimmune encephalopathy was suggested by Brain and colleagues in 19667 and was subsequently confirmed by other authors. The majority of reported patients were euthyroid at the time of neurological presentation, and alteration in the thyroid metabolic status clearly did not explain the encephalopathy in these patients. Hashimoto’s encephalopathy, also called steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT), is characterized by subacute onset of altered level of consciousness, cognitive impairment, stroke-like episodes, and focal or generalized seizures. There is a preponderance of female patients with a female to male ratio of 10:1, and the course of the disease is usually relapsing that should alert the physician to the diagnostic possibility of Hashimoto’s encephalopathy.8 Hypothyroidism is a common disorder and anti-thyroid peroxidase antibodies can occur in the general population. Therefore, the diagnosis of Hashimoto’s encephalopathy may be challenging due to the lack of causality and the possible coincidence of separate diseases. In addition, many other antibodies (most notably anti-mGluR1 antibodies) can be found in patients with autoimmune ataxia. If extensive antibody screening has not been made in the CSF, the presence of autoantibodies more specific than anti-thyroid peroxidase antibodies cannot be excluded.
Thyroid peroxidase and thyroglobulin, the main thyroid autoantigens, are the enzyme-substrate pair involved in thyroid hormone production.9 Thyroid peroxidase is a 933-amino-acid, which carries out the iodination and the intramolecular coupling of tyrosine residues of thyroglobulin to form thyroid hormones. Several thyroid-related auto-antibodies may react against these autoantigens with subsequent development of widespread systemic manifestations.10 It is estimated that around one-fourth of patients with Hashimoto’s thyroiditis have associated systemic disease. These include pernicious anemia, myasthenia gravis, Addison’s disease, and ulcerative colitis.11
Immune-mediated cerebellar ataxia due to anti-peroxidase antibody has been associated with autoantibodies to cerebellar structures, specifically, cerebellar astrocytes expressing glial fibrillary acid protein. The exact epitope that causes cross-reaction is not yet identified.12 In that study, the anti-thyroid peroxidase antibodies from patients’ CSF reacted with monkey astrocytes, while anti-thyroid peroxidase antibodies from patients with thyroiditis and no neurological symptom did not. This may suggest that either anti-thyroid peroxidase antibodies from Hashimoto’s encephalitis patients are somewhat different from the usual anti-thyroid peroxidase antibodies. It may also suggest that there is actually another antibody involved. Further basic science research is required to address these questions. Studies with reported patients having a high level of anti-thyroid peroxidase showed a good response to steroids, but in these cases, the level never reached 700. In our case, the anti-thyroid peroxidase level was very high (more than 1000), which may explain little or no response to steroid therapy, and aggressive therapy such as intravenous immunoglobulins or plasma exchange should be used on top of steroid therapy. The presence of normal thyroid function and very high autoantibodies have excluded hormonal deficiency or alteration as the etiology. Other features that argue in favor of autoimmune ataxia include young female patient, acute and subacute progression, and good response to immunomodulation.
4-aminopyridine (prolonged-release fampridine) is a lipid-soluble selective potassium channel blocker that readily crosses the blood-brain barrier and is currently indicated for walking improvement in adult patients with multiple sclerosis.13 It acts on the surface of nerve fibers to reduce the leakage of ionic current from potassium channels in demyelinated axons, thereby inhibiting repolarization and prolonging the duration of action potentials, presumably allowing for more action potential propagation along the cell membrane.14 This medication is used off-label for walking improvement in patients with hereditary ataxia with success in some patients.15 This is a single case report, and although our experience suggests that this medication is not effective in the management of autoimmune cerebellar ataxia, large scale studies should be done for an accurate recommendation.
Conclusion
Early diagnosis of Hashimoto’s encephalopathy with autoimmune cerebellar ataxia and intervention with immunomodulatory therapy are of paramount importance. Close monitoring after steroid therapy is important since some patients with this rare disease might be resistant to steroid therapy and require aggressive immunomodulatory therapy. The presence of cerebellar atrophy and high levels of anti-thyroid peroxidase (more than 1000) may suggest the need to start aggressive therapy with steroids and other immunomodulatory therapy such as intravenous immunoglobulins or plasma exchange. The presence of cerebellar atrophy should not prevent the treating team from being aggressive in the management of this disease. 4-aminopyridine is not effective in the management of autoimmune cerebellar ataxia. However, more studies need to be done for this rare disease especially those which carry longitudinal data of several cases instead of one case report. Clinico-laboratory correlation (clinical improvement with the magnitude of autoantibody titer) is important.
Clinical Practice Guidelines
Clinical Practice Guidelines must include a short abstract. There should be an Introduction section addressing the objective in producing the guideline, what the guideline is about and who will benefit from the guideline. It should describe the population, conditions, health care setting and clinical management/diagnostic test. Authors should adequately describe the methods used to collect and analyze evidence, recommendations and validation. If it is adapted, authors should include the source, how, and why it is adapted? The guidelines should include not more than 50 references, 2-4 illustrations/tables, and an algorithm.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received March 3, 2019.
- Accepted May 15, 2019.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.