


Abstract
Objectives: To describe the clinical phenotype of eight children diagnosed with CD59 deficiency and their ultimate neurological outcome.
Methods: The data of our cases were extensively reviewed both clinical and ancillary tests; investigations included: neuroimaging, neurophysiological studies, and laboratory tests.
Results: All patients presented during early infancy with Guillain-Barre syndrome later they suffered repeated relapses leading to the diagnosis of chronic axonal neuropathy. Recurrent stroke and acute necrotizing encephalopathy were described, 2 patients in each group. One girl developed acute disseminated encephalomyelitis while one boy developed acute transverse myelitis. Overt hemolytic anemia requiring blood transfusion reported in six patients.
Conclusion: Inherited CD59 deficiency is an autosomal recessive disorder which can have devastating neurological consequences. First line immunotherapy including intravenous immunoglobin, corticosteroids, and plasma exchange may have transient beneficial effect. Reports of targeted therapy with eculizumab might be lifesaving. Genetic counseling is crucial.
Inherited CD59 deficiency is a rare autosomal recessive disorder manifesting during early childhood with incidence of less than one per million.1 The CD59 deficiency presents during early infancy with protean manifestations including chronic hemolysis and peripheral demyelinating disease mimicking recurrent Guillain-Barre syndrome (GBS) or chronic inflammatory demyelinating polyneuropathy (CIDP).2 The CD59 orchestrates the complement cascade by inhibiting the formation of membrane attack complex (MAC).3 Patients with deficient CD59 may suffer disinhibited complement amplification following viral or post viral infection.3 The expression of CD59 in normal human nervous system was revealed at different levels including Schwann cells, neurons, endothelial cells, microglia, oligodendrocytes, and astrocytes.4 Revising brain autopsy of unsolved disease in a child who developed recurrent brain infarction with retinal and optic nerve involvement, CD59 protein expression was missing on brain endothelial cells by immunohistochemical staining.5 We describe multiple cases with CD59 deficiency presented with chronic axonal neuropathy, cerebrovascular disease, and chronic Coombs negative hemolysis. Recognition of this condition may help to draw management pathway and in family counseling.
Methods
In this case series we have identified 8 children diagnosed with inherited CD59 deficiency at Salmaniya Medical Complex, a tertiary hospital in Bahrain. The clinical manifestations and different presentations were reviewed. The diagnostic assessments and therapeutic interventions for each of the cases were studied.
Results
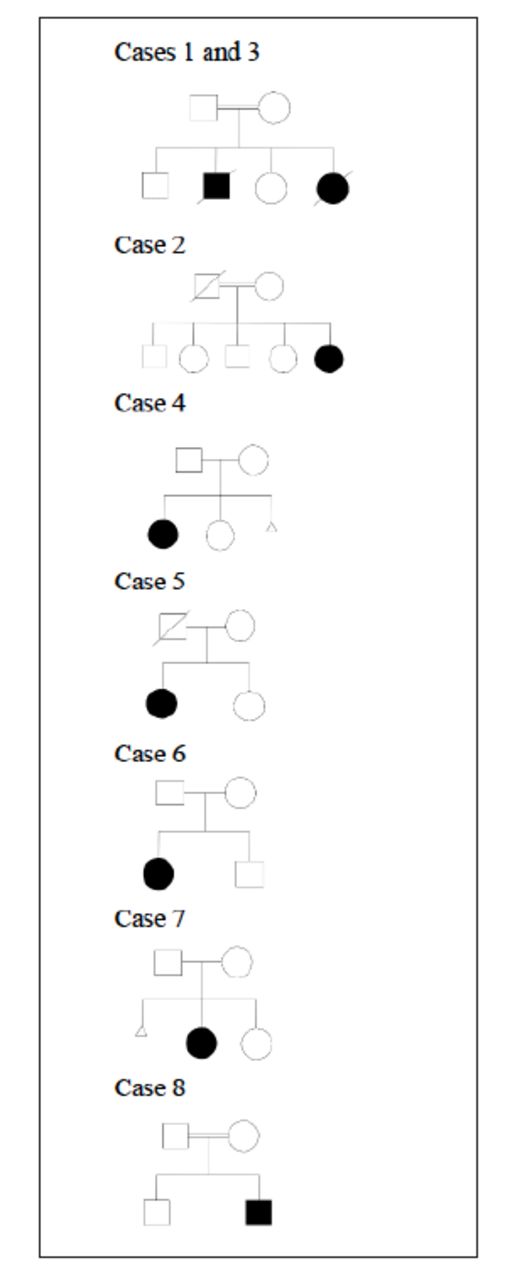
Six girls and 2 boys were reported, all of them presented during infancy ranging from 2 months up to 11 months. Four children were of consanguineous marriages, and 5 families originated from the same island (Sitra). Figure 1 illustrates their pedigrees.
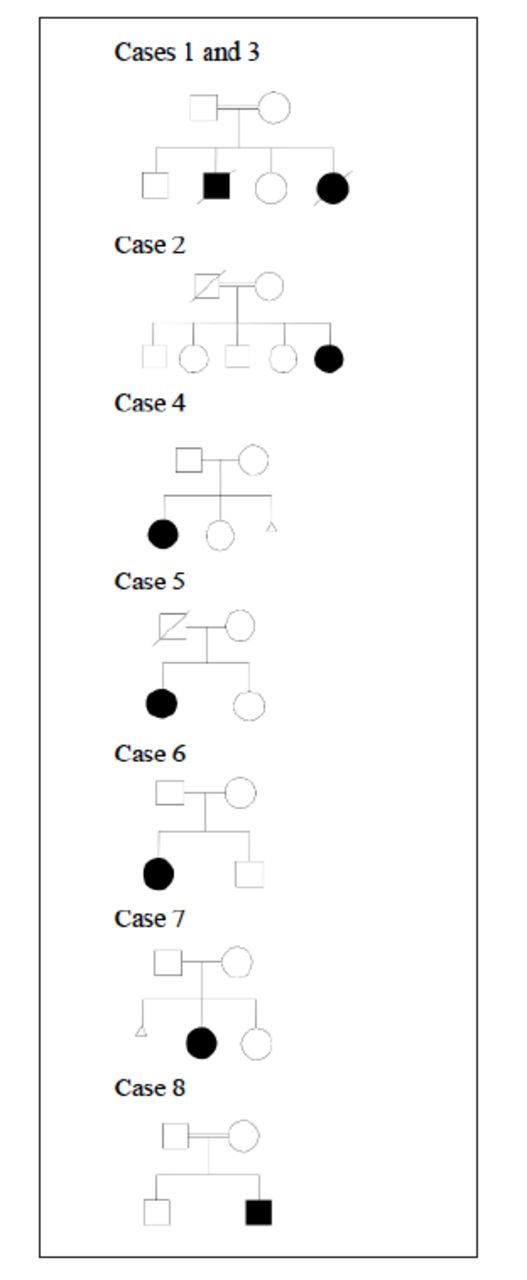
- Pedigree of families of identified patients
All children had viral prodrome, low grade fever or recent vaccination prior to their presentation at times triggering relapses. Acute flaccid paralysis with profound hypotonia and hyporeflexia was observed in all cases at their initial presentation except one case which was associated with acute encephalopathy. Seven patients continued to have recurrent relapses of weakness and developed the phase of recurrent axonal polyneuropathy.
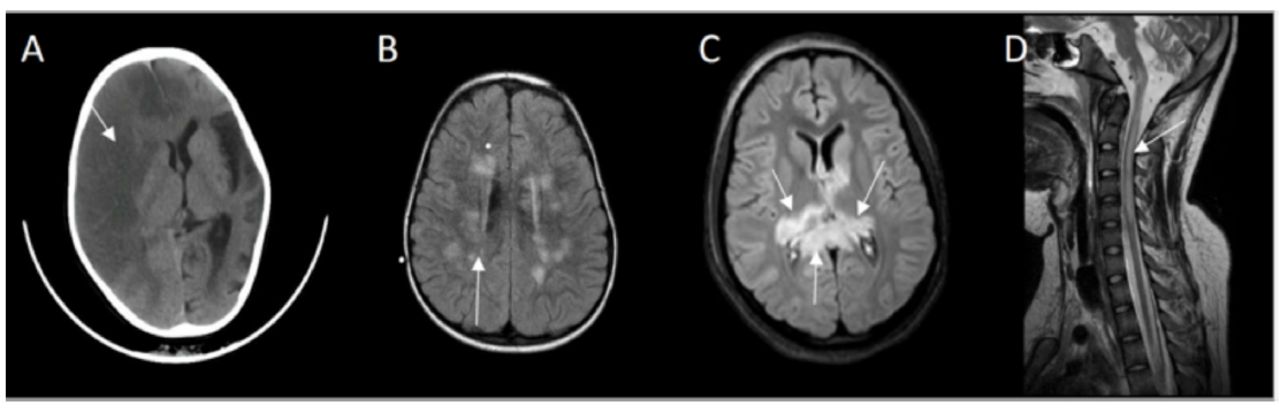
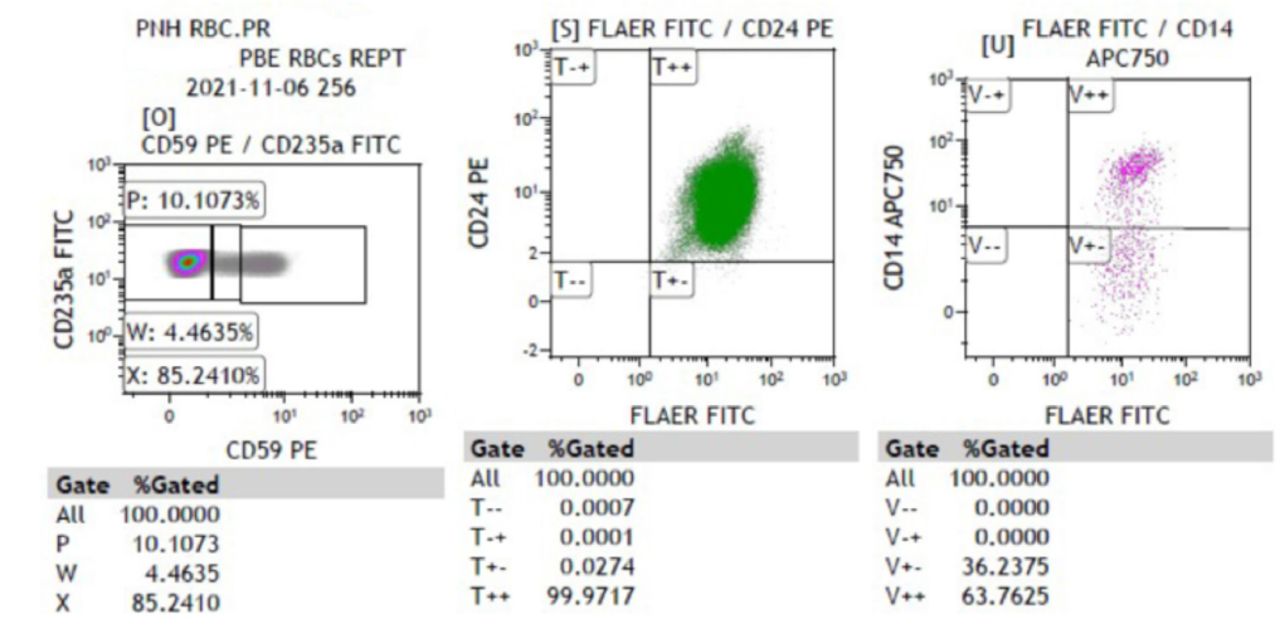
Four patients demonstrated symptoms of angioedema with facial swelling requiring intubation in one case due to laryngeal edema. Guillain-Barre syndrome and recurrent axonal neuropathy were treated with either intravenous immunoglobin, corticosteroids or plasma exchange. The responses were incomplete, and recoveries were not optimal. They continued to accumulate more neurological deficits following each relapse. Case number six developed recurrent axonal neuropathy and one episode of acute disseminated encephalomyelitis, she had the opportunity to receive eculizumab but showed no meaningful response. Extensive investigations were conducted according to their presentation and plausible differential diagnosis. Nerve conduction studies revealed either axonal or demyelinating sensory motor neuropathy. Computed tomography (CT) or Magnetic resonance imaging (MRI) of the brain and spine was abnormal in 5 cases, and showed either stroke, acute disseminated encephalomyelitis (ADEM), acute necrotizing encephalitis (ANE) or acute transverse myelitis (Figure 2A-2D). Eventually they were diagnosed either by flowcytometry (Figure 3) or genetic testing.
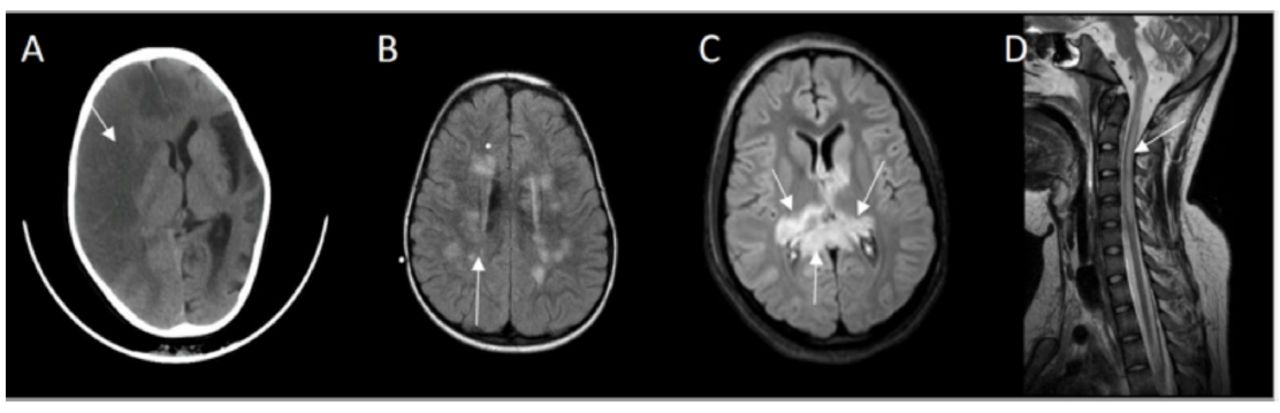
- CT brain showing A) Case 7: right hemispheric loss of gray-white matter interface and effacement of sulci affecting right anterior and middle cerebral artery territory, midline shift and left hemispheric cortical atrophy. B) Case 6: FLAIR MRI depicting multiple high signal intensity lesions in centrum semiovale and periventricular white matter. C) Case 4: FLAIR MRI revealed bilateral asymmetrical high signal intensities of the thalami and splenium of corpus callosum. D) Case 1: T2 MRI midline sagittal showing high signal intensity extending from medulla oblongata up to C5 of cervical cord.

- Flow cytometric analysis of case 8 shows a PNH clone within RBC (89.7%), the findings suggestive of paroxysmal nocturnal hematuria. RBC - Red blood cells, PNH - Paroxysmal Nocturnal Hemoglobinuria, FITC - Fluorescein isothiocyanate
A tailored NGS panel CentoNeuro™ (sequencing including NGS-based CNV analysis) was performed for 8 patients. The DNA was fragmented using enzymes and then enriching of regions of interest was performed using DNA capture probes. Illumina platform was used to sequence the final indexed libraries. The coding regions of the panel genes, 10 base pair of flanking intronic sequences, and known pathogenic/likely pathogenic variants within these genes (coding and non-coding) were targeted for analysis.
All 8 patients tested were homozygous for the same mutation c.323C>A in the CD59 gene (NM_000611.5:c.323C>A), p.(Ser108). The CD59 gene is located on chromosome 11p13 and this mutation is known to cause a premature stop codon. All disorders linked to this gene are associated with loss of function. The clinical features and diagnostic tests were summarized in Tables 1 & 2.
- Clinical manifestations.
- Diagnostic test and outcome.
Upon follow up, 2 patients developed recurrent arterial ischemic stroke one of them developed massive right middle cerebral artery stroke requiring decompressive craniectomy. Two patients developed features of acute necrotizing encephalopathy and seizures. One girl experienced acute disseminated encephalomyelitis. Case number 1 developed at one stage acute transverse myelitis leading into quadriparesis requiring permanent ventilation. The CNS is spared in three cases so far. None of them had hemolytic anemia as initial presentation, it was discovered in retrospect or developed gross hematuria at one stage requiring blood transfusion.
Discussion
Paroxysmal nocturnal hemoglobinuria (PNH) is frequently associated with secondary CD59 deficiency, while primary CD59 deficiency due to germline mutation is extremely rare.2 Certain neurological manifestations are described with congenital CD59 deficiency such as demyelination, Guillain-Barre syndrome and stroke.6 This syndrome is often triggered by common febrile illness in infancy and relapse might occur with immunization. A 128-amino acid GPI-anchored cell surface glycoprotein is encoded by CD59 gene that is critical for protein function. CD59 is also known as protectin as it protects the cells from complement mediated lysis by inhibiting the final step of membrane attack complex (MAC).4 Mutation in Cys89Tyr is associated with improper localization of CD59 on cells surface clinically manifesting as chronic hemolysis in infancy with peripheral demyelinating diseases due to myelin and axonal damage.2 The CD59 deficiency is a common finding in patients with paroxysmal nocturnal hemoglobinuria (PNH) especially adults.7 Case number eight presented with hemolysis evidence by hematuria and confirmed diagnosis of PNH by flow cytometry (Figure 3). All of our children were homozygous for this mutation c.323C>A p.(Ser108). This variant has previously been described as disease causing for hemolytic anemia, CD59-mediated, immune mediated polyneuropathy by Weinstock.8 It is classified as likely pathogenic (class 2) according to the recommendations of American College of Medical Genetics (ACMG).9
A very severe complication of the disease is thrombosis affecting 29 to 44% of patients.10 This might occur in unusual sites such as portal circulation, spleen, liver, skin, or brain. Case number 7 presented with right middles artery stroke and left hemispheric atrophy (Figure 2A).
The disease has an inflammatory nature suggested by high protein level in the cerebral spinal fluid and thickened hyperintense areas on neuroimaging. Some cases on the other hand (N=3) had normal brain MRI or CT scans. Of the patients who underwent CSF analysis, CSF proteins is observed to be high in variable degrees.
Solmaz et al11 has reported the same variant as the cause of recurrent demyelinating episodes in one of his Turkish patients affected with inherited CD59 deficiency. His patient presented at 1 year with convulsions, hemiparesis and paralysis of cranial nerves. The MRI showed thalamic, cerebellar, temporal, spinal lesions.
While in this case report all of our patients had their diagnosis confirmed by DNA sequencing, the founder effect is clearly pronounced as all patients originated from the same geographical area where it used to be a relatively isolated island with a high consanguinity rate during last century. The high incidence as compared to the total population number of Bahrain also support the founder effect theory.
Nevo et al1 has reported homozygous missense mutation p.Cys89Tyr in CD59 gene, in 5 children suffered from chronic Coombs-negative and relapsing neuropathy while central nervous system was not involved and the cognitive function was intact;1 while Haliloglu et al12 describe 3 children presented with strokes, chronic immune-mediated peripheral neuropathy and hemolysis due to homozygous missense mutation p.Asp49Val in the CD59 gene mimicking the presentation of our cases.
It is noted that prognosis is variable depending on the initial neurological deficit that can be fatal or possess significant morbidity. Eculizumab might be effective in treating the neurological symptoms of inherited CD59 deficiency to possibly complete resolution.7 In PNH, Eculizumab therapy causes rapid reduction in the intravascular hemolysis with corresponding reduction in lactate dehydrogenase levels in some of the cases.7 In our series one case received Eculizumab in addition to treatment with intravenous immunoglobulins (IVIg) and corticosteroids. The patient had poor outcome mostly due to extensive demyelination of the supra and infratentorial structures including brainstem following acute disseminated encephalomyelitis.
Conclusion and future directions
Genetic counseling is imperative not only for parents as it may be valuable to the extended family as well. Performing haplotype analysis to confirm the founder effect can be of beneficence. We recommend genetic testing for the parents to determine the carrier status and to confirm homozygosity in place of compound heterozygosity for a large deletion. The ability to develop rational therapy is dependent on an improved basic understanding of the mechanism by which CD59 regulates the immune system. Future targeted therapies will likely be obtained from detailed knowledge of molecular and cellular biology of the tissue involved in CD59 deficiency and consequences of CD59 gene loss on these cell lines.
Acknowledgment
We would like to thank the teams of neurosciences, genetics and pathology at Salmaniya Medical Complex for their efforts in the diagnosis and management of the patients and the contribution to having this publication. We would like to thank as well Editage (editage.com) for English language editing.
- Received October 12, 2022.
- Accepted February 8, 2023.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.