


Abstract
Objectives: To evaluate the clinical and electrographic features of patients with autoimmune epilepsy and assess the influence of early diagnosis and treatment on reducing seizure frequency.
Methods: A retrospective observational case series was conducted utilizing medical records from King Abdullah Medical Hospital between 2017 and 2022. Cases of newly diagnosed seizures were chosen based on laboratory-proven autoimmunity.
Results: Five female inpatients were identified, primarily presenting with seizures suggestive of an autoimmune origin. Autoimmune antibodies were detected in all patients as follows: GAD (3), NMDA-R (2). One patient exhibited unilateral temporal lobe onset while three displayed bilateral onset. One patient had an associated malignancy. Rituximab was administered as an immunomodulatory therapy to four patients, resulting in successful seizure reduction post-immunotherapy initiation.
Conclusion: Autoimmune epilepsy is recognized as a distinct condition. The clinical presentation can be complex and antibody testing may warrant repetition if initial results are negative or if specific antibodies are not detected. Early initiation of immunosuppression, coupled with prompt treatment escalation when required, is vital for achieving optimal patient outcomes.
Autoimmune epilepsy is an underrecognized condition lacking standardized guidelines for management. It is crucial to diagnose an autoimmune cause for epilepsy, as such patients may remain refractory to conventional antiepileptic medications.1 Research indicates that approximately 10% of epilepsy cases are attributed to autoimmune origins.2 Potential clinical presentations include an acute or subacute onset, a high seizure frequency, intra-individual seizure variability or multifocality, resistance to antiepileptic drugs, personal or familial history of autoimmunity, and a record of recent or past neoplasia.3
Important diagnostic tools such as cerebrospinal fluid and serum analysis for the detection of neural-specific autoantibodies can facilitate the establishment of an accurate diagnosis and inform treatment strategies. Additional supportive investigations include detecting inflammation via magnetic resonance imaging or fluorodeoxyglucose positron emission tomography (FDG-PET).3
Several autoantibodies have been associated with autoimmune epilepsy, including those targeting neuronal intracellular antigens or neuronal cell surface antigens. The latter category includes antigens like the voltage-gated potassium channel (VGKC) complex, NMDA and AMPA glutamate receptors, and GABA receptors, while the intraneuronal autoantibodies include anti-Hu, anti-Ma2, and anti-CRMP-5.1
In this study, a comprehensive review was conducted on 5 recent cases of autoimmune epilepsy admitted to a tertiary hospital’s neurology department.
Analyses were performed on patient data, including age, gender, clinical manifestations, brain MRI, EEG, CSF test results, treatment protocols, and treatment efficacy. The insights gleaned from this analysis aim to augment disease understanding, thereby facilitating earlier diagnosis and intervention.
Case Report
A 24-year-old right-handed female with a history of diabetes mellitus presented at our hospital with intractable epilepsy. At the age of 14 years, the patient had her first seizure, which included generalized tonic-colonic convulsions, loss of consciousness, upward ocular deviation, and urinary incontinence. She had no history of trauma, inflammation, infection, psychiatric disease, or medication use. Moreover, she had no relevant risk factors for epilepsy, and her family history was unremarkable.
Despite being started on 3 antiepileptic medications, the patient’s seizures remained uncontrolled. She was admitted to the Epilepsy Monitoring Unit (EMU) in our hospital for seizure classification and diagnosis. Subsequent video monitoring identified seizures with bilateral hand automatism, which were associated with coughing and chewing, followed by generalized tonic-clonic seizure, postictal drowsiness, and fatigue, with no preictal aura. Interictal EEG revealed multiple independent bitemporal sharp waves, prominent left temporal and left frontal spike and waves, with ictal EEG of 2 recorded seizures initiating from the right temporal lobe with subsequent generalization, and one seizure originating from the left frontal lobe.
Complete blood workup was performed, including routine assessments of electrolytes and hepatic and renal functions. All values were within normal parameters, with the exception of an elevated serum level of GAD 65 at >2000 IU/ml. CSF analysis was largely normal, apart from an elevated anti-GAD 65 level (>2000 IE/ml).
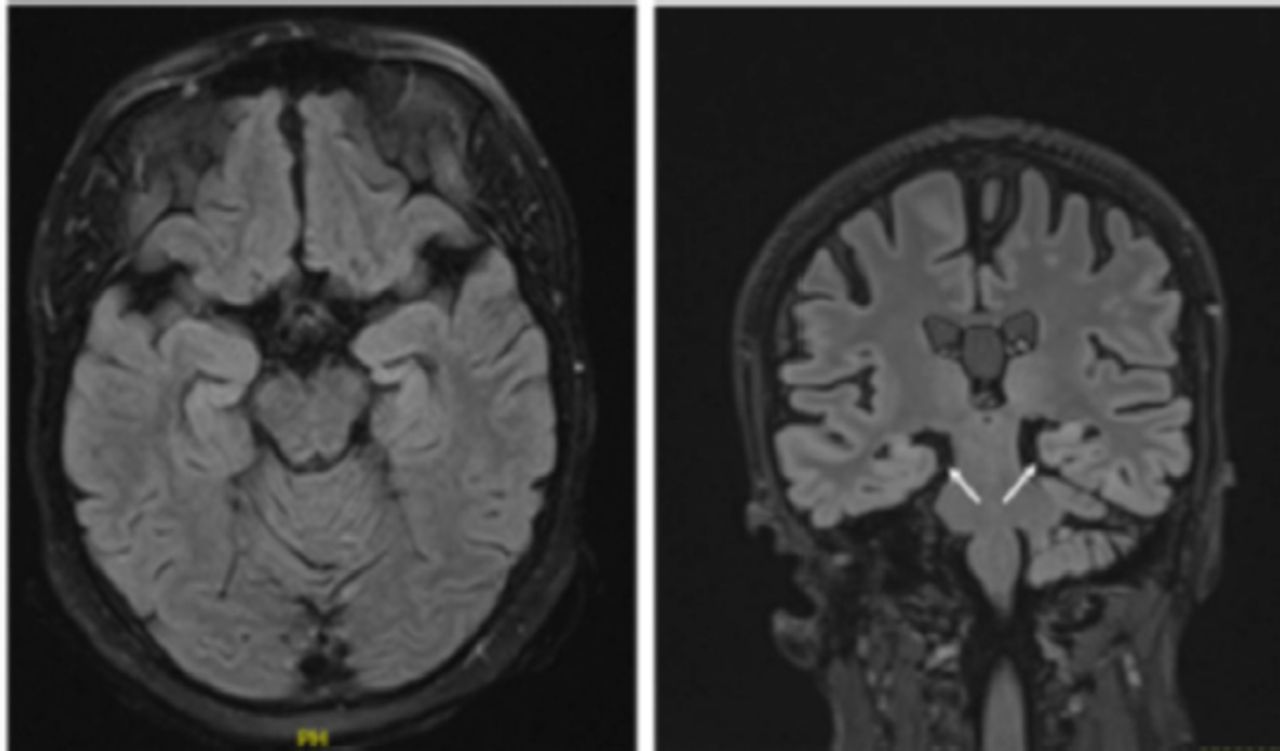
Brain magnetic resonance imaging (MRI) revealed bilateral mesial temporal sclerosis, which was more pronounced on the right side (Figure 1). A fluorodeoxyglucose (FDG) positron emission tomography (PET) scan revealed severe right temporal and mild-to-moderate left temporal hypometabolism.
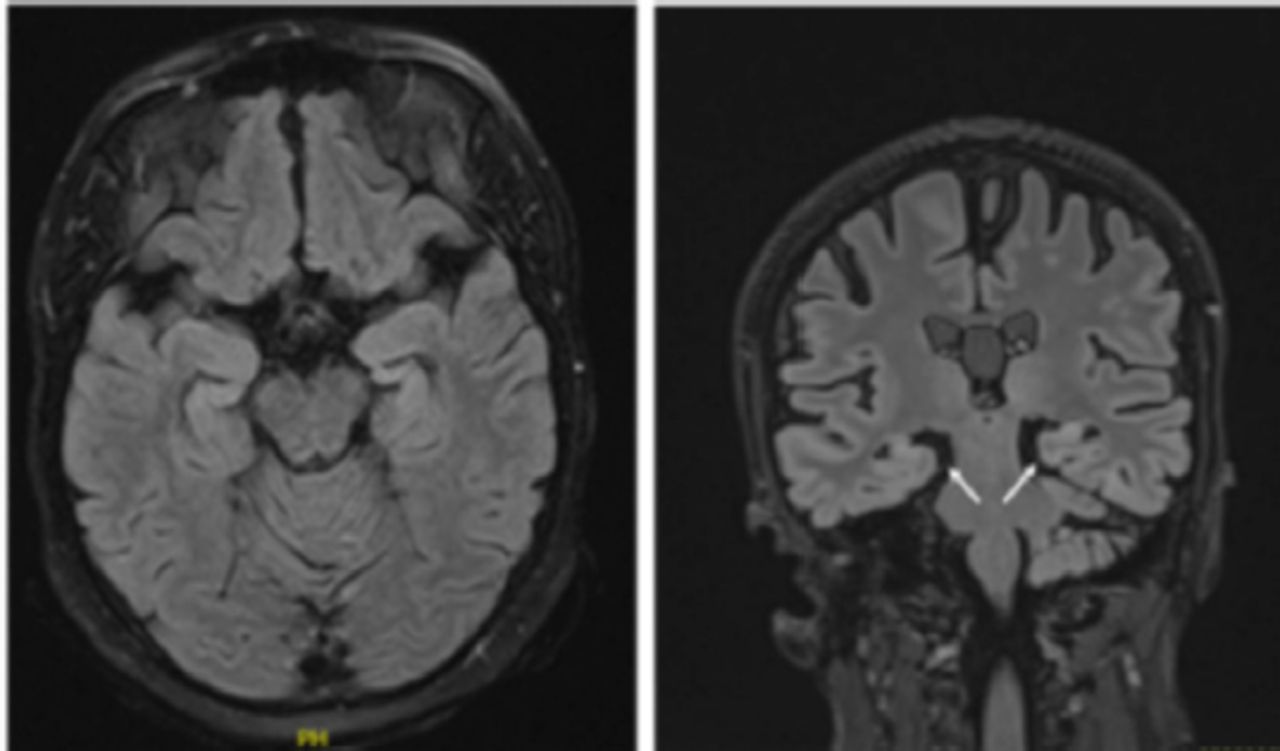
- Brain magnetic resonance imaging (MRI) demonstrating abnormal bilateral hippocampal volume loss more pronounced on the right side.
The CT scans of the chest, abdomen, and pelvis and transvaginal ultrasound were unremarkable without evidence of neoplasm or ovarian teratoma.
The patient was initially administered an oral dose of prednisolone (60 mg daily) for one week, which was subsequently followed by intravenous rituximab treatment (1,000 mg biannually). Given her diagnosis of intractable epilepsy, she was prescribed a combination of clobazam (10 mg twice daily), levetiracetam (750 mg twice daily), and lacosamide (200 mg twice daily) for seizure management.
The most recent clinic follow-up occurred this month, during which it was documented that seizure control was maintained with the original regimen of antiepileptic medications, albeit with a revised dose of levetiracetam (increased to 1,500 mg twice daily) and the ongoing biannual rituximab treatment.
Case 2
A 19-year-old right-handed female was referred to our hospital for refractory status epilepticus. Ten days prior to presentation to the hospital, she reported abdominal pain, vomiting, and fever. On subsequent days, she developed sudden left-sided weakness, numbness, intermittent confusion, and multiple episodes of generalized tonic-clonic seizures. The initial neurological examination revealed a severely ill patient with generalized body twitching and neck stiffness. Her pupils, each 2 mm in diameter, were reactive to light. Further examination showed left-sided hypotonia and brisk tendon reflexes, with a muscle power rating of 4+/5 on the right side, 2/5 on the left leg, and 3/5 on the left arm.
An initial brain CT scan and comprehensive blood workup were unremarkable. Additional analysis revealed a mildly elevated total CSF protein level (48.9 mg/dl), while cultures, Gram stain, and HSV PCR were negative. The patient was admitted to the intensive care unit and was started on levetiracetam and sodium valproate. Initially, a presumptive diagnosis of meningoencephalitis led to treatment with acyclovir, ceftriaxone, vancomycin, ampicillin, and dexamethasone, but her condition showed no improvement. Due to refractory status epilepticus necessitating intubation, video-recorded EEG monitoring was implemented while under midazolam and propofol sedation and continued for three consecutive days. The EEG revealed no clinical or subclinical seizures but showed slow sharp waves in the right hemisphere.
- Clinical characteristics.
A comprehensive history provided by the family revealed that the patient had a previous incidence of seizures 4 years ago and a history of a psychiatric condition that was treated with unspecified psychiatric medication for a duration of 18 months.
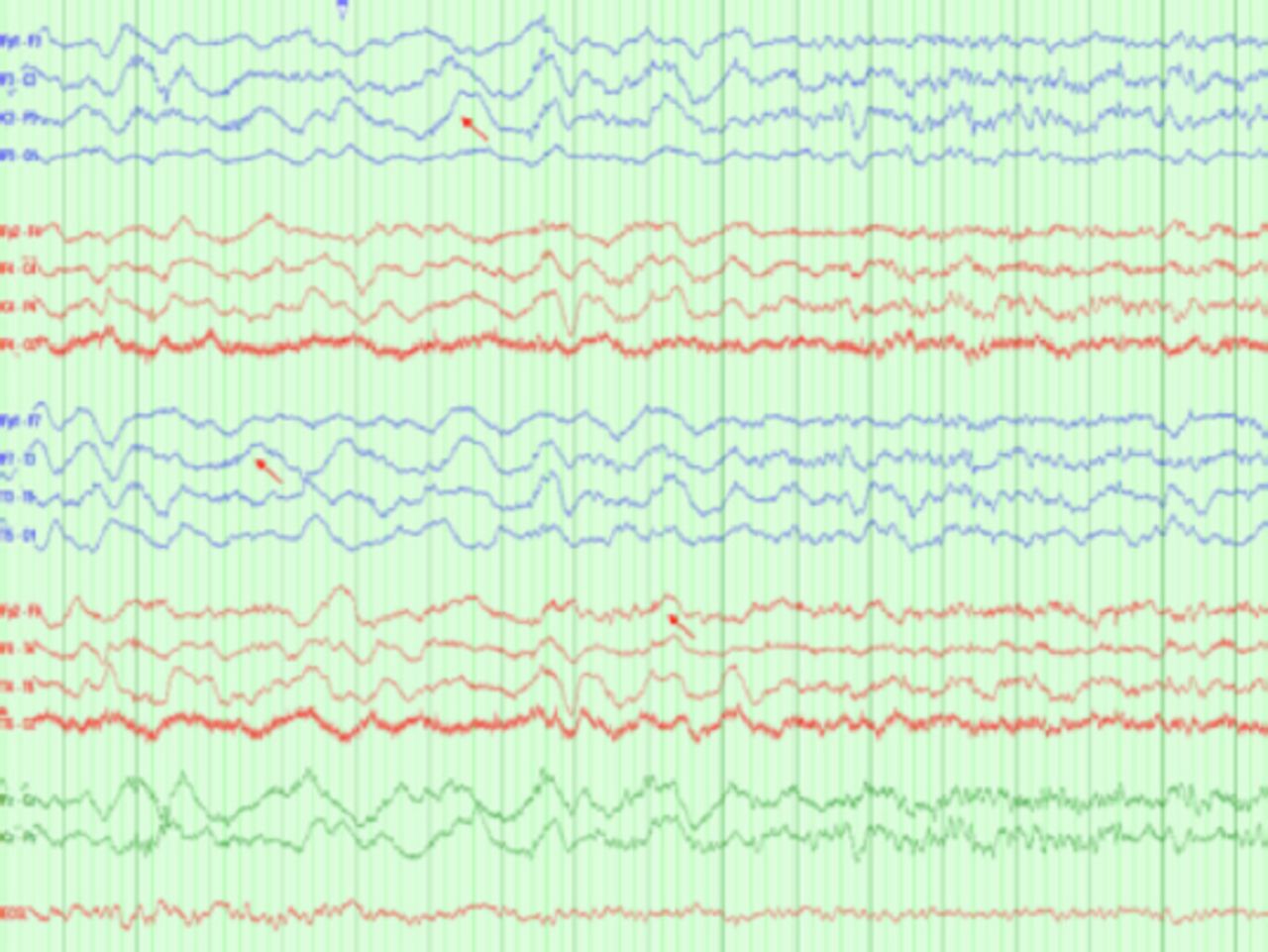
Upon discontinuation of the sedatives, the patient exhibited orofacial dyskinesia-like movements but no seizures. Notably, a subsequent EEG recording demonstrated a delta brush pattern (Figure 2).
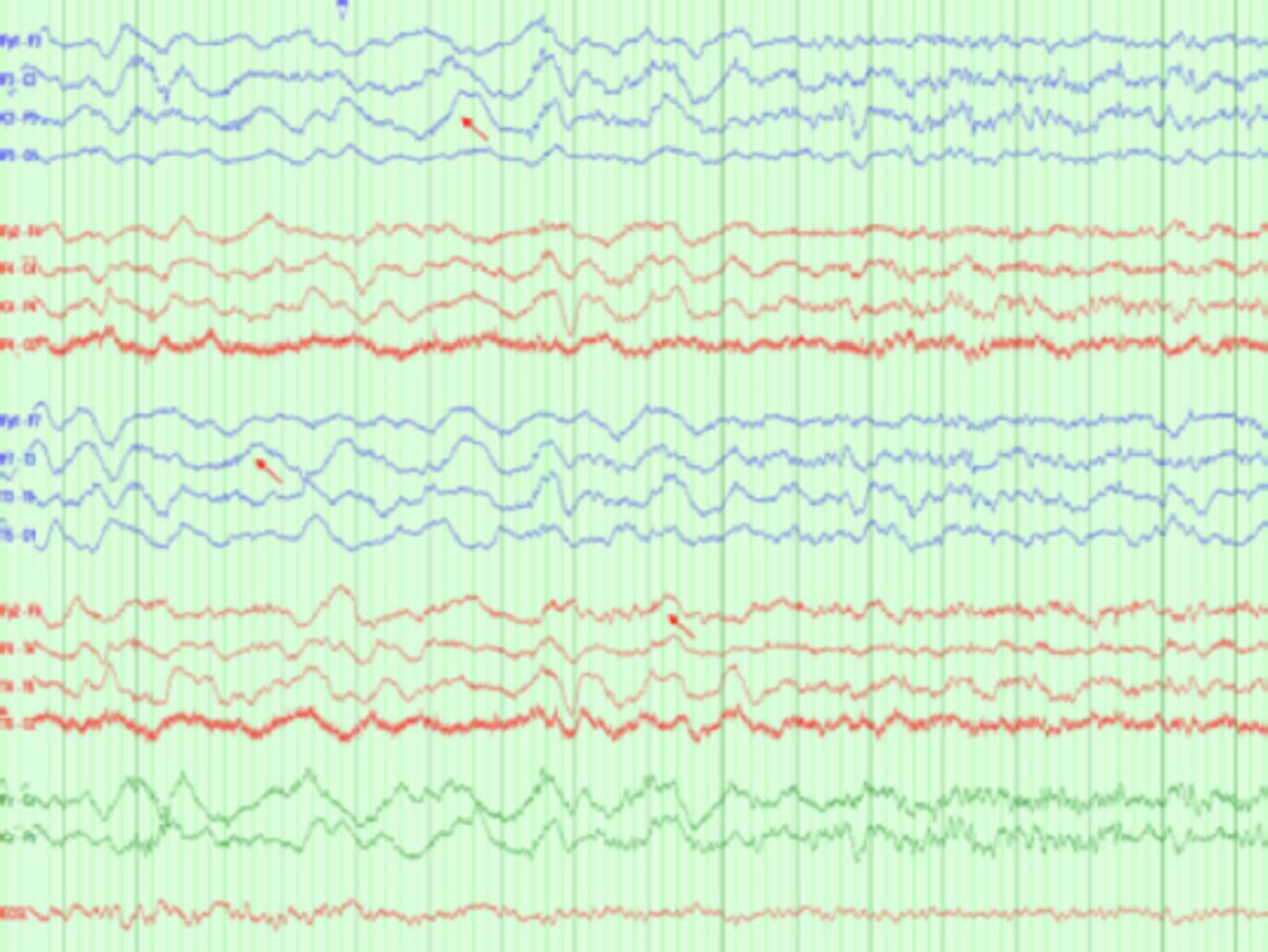
- Interictal EEG showing diffuse slowing with a delta brush, a pattern characteristic of autoimmune encephalopathy associated with Anti-NMDA antibody.
The diagnosis of autoimmune encephalitis was eventually established due to the patient’s unresponsiveness to AEDs and the presence of inflammatory CSF. The therapeutic approach comprised a five-day regimen of intravenous methylprednisolone at 1,000 mg, 7 plasma exchange sessions, and a series of 6 IVIG infusions. This regimen significantly mitigated seizure frequency, albeit without ameliorating psychiatric symptoms. Furthermore, MRI of the brain yielded no notable findings.
A subsequent lumbar puncture and autoimmune panel unveiled the presence of anti-NMDA receptor antibodies. Both CT scans of the chest, abdomen, and pelvis, as well as a transvaginal ultrasound, showed no indications of neoplasms or ovarian teratomas.
After achieving seizure control via a three-AED regimen and intravenous rituximab at 1,000 mg biannually, the patient was discharged.
During a follow-up period of 18 months, she exhibited progressive improvement in psychiatric symptoms and remained seizure-free. The current medication regimen includes levetiracetam at 750 mg twice daily and rituximab.
Case 3
A 53-year-old right-handed female with a history of diabetes mellitus, hyperthyroidism, and mature cystic teratoma who was scheduled for a total hysterectomy was referred by the anesthesia department due to episodes of altered behavior and loss of consciousness. These episodes, consisting of abnormal limb jerks, had been first noticed 20 years prior, typically preceded by an onset of fear and occasionally resulting in tongue biting. The episodes were particularly provoked by music. She was initially started on carbamazepine without achievement of seizure control.
For a more accurate diagnosis and seizure classification, the patient was admitted to the EMU at our hospital. Continuous video monitoring revealed seizure semiology of behavioral arrest that started with bimanual automatism and concluded with postictal fatigue and confusion. During the ictal recording, 3 focal seizures were identified: one in the right frontotemporal region and two in the left frontotemporal region. Notably, the patient’s speech remained unaffected during the right temporal seizure.
Interictal EEG showed intermittent rhythmic sharp waves predominantly in the right bitemporal area. Comprehensive blood work, including routine tests for electrolytes and liver and renal functions, were within normal ranges. The CSF analysis was largely unremarkable, with the exception of a significantly elevated level of anti-glutamic acid decarboxylase 65 (>2,000). Concurrently, brain MRI and CT scans of the chest, abdomen, and pelvis did not reveal any abnormalities, other than a large adnexal cystic lesion (cystic teratoma).
Following these investigations, the patient underwent a total abdominal hysterectomy with bilateral salpingo-oophorectomy and omentectomy. Afterward, she was initiated on a regimen of rituximab, with a dose of 1,000 mg intravenously every 6 months.
In over 2 years of follow-up at our clinic, the patient achieved seizure control on a regimen comprising levetiracetam 250 mg twice daily, lacosamide 100 mg twice daily, and rituximab.
Case 4
A 38-year-old right-handed female was referred to our hospital for refractory status epilepticus and epilepsia partialis continua. She first reported seizure symptoms, characterized by facial twitching, 4 years prior and had also been experiencing cognitive alterations and short-term memory impairment over the previous year. Upon admission to our critical care unit, the patient was subjected to continuous video EEG. Initial 24-hour EEG data revealed frequent subclinical seizures localized to the right temporal lobe along with numerous clinical focal seizures. Prior to transfer to our hospital, the patient had been prescribed multiple AEDs. Sedation (with midazolam and propofol infusion) and intubation were necessitated to maintain EEG burst suppression.
Results from comprehensive bloodwork, including routine tests for electrolytes, liver, and renal function, were within normal ranges. Brain MRI demonstrated the presence of right mesial temporal sclerosis.
A lumbar puncture was performed for CSF analysis to evaluate the possibility of autoimmune epilepsy. Seizure control was achieved after 7 days of continuous sedation. However, 2 days later, the patient experienced a resurgence of clinical and subclinical seizures accompanied by aggressive behavior and other behavioral abnormalities.
Considering the potential diagnosis of autoimmune epilepsy and the patient’s concurrent right ear and chest infections, a regimen of plasma exchange (5 sessions) and pulse steroids (5 days) was implemented following the resolution of the infections. This was followed by daily administration of 60 mg of oral prednisolone.
Upon clinical and electrographic improvement, the patient was transferred to the EMU for long-term EEG monitoring. Here, ictal EEG recorded independent bitemporal epileptiform discharges, predominantly on the right side, with no seizures recorded. Subsequent CSF examination revealed positive anti-glutamic acid decarboxylase 65 (>2,000), with no other abnormalities detected.
Rituximab therapy was initiated with a dosage of 1,000 mg intravenously every 6 months. During the subsequent 3-year clinic follow-up, the patient achieved seizure control with sodium valproate (1,000 mg twice daily), lacosamide (150 mg twice daily), and the rituximab regimen.
Case 5
A 35-year-old right-handed female with a history of migraines and Crohn’s disease, the latter managed with adalimumab injections and 50 mg azathioprine daily, was referred to the Neurology Clinic for seizure management.
The patient began experiencing generalized tonic-clonic seizures 20 years prior, each episode persisting for several minutes. Each seizure was preceded by feelings of fear, followed by postictal drowsiness and generalized body fatigue. The patient was initiated on a regimen of carbamazepine, and upon the addition of lacosamide, seizure control was achieved.
The patient was admitted to the EMU in our facility for seizure classification and diagnosis. No seizure activity occurred during her admission. However, her interictal EEG showed independent bitemporal sharp waves.
A comprehensive blood panel was performed, including routine evaluations of electrolytes, liver, and renal functions. All results fell within normal parameters except for the serum epilepsy autoimmune panel, which showed an elevated level of glutamate receptor antibody (type NMDA) >1:40. The CSF analysis was unremarkable.
Brain MRI revealed a few nonspecific subcortical and deep white matter T2 FLAIR hyperintense foci involving bilateral frontal lobes, the left external capsule, and the left temporal pole.
Upon follow-up in the epilepsy clinic, the patient achieved seizure control with a regimen of 400 mg carbamazepine twice daily.
Discussion
Refractory epilepsy often poses a formidable diagnostic challenge, resulting in recurrent, prolonged hospitalizations and ICU stays. Recently, autoimmune epilepsy has been recognized as a potential treatable cause of intractable epilepsy. Its first documentation dates back to 1960 as an autoimmune encephalitis associated with epilepsy.4 It was subsequently defined as a subset of autoimmune encephalitis typically unresponsive to standard antiepileptic drugs.5 Key features of this condition include seizure resistance to antiepileptic drugs, high seizure frequency, and a possible medical or familial history of autoimmune disease or malignancy.6
In our study, we evaluated 5 patients who experienced high-frequency refractory seizures resistant to conventional antiepileptic drugs. Interestingly, none of the patients exhibited a medical or familial history of autoimmune diseases, with one exception of a patient with a prior malignancy.
A previous retrospective study suggested that new-onset refractory seizures can transpire at any age and in the absence of any discernible triggers.7 Parallel to this finding, in our cohort, four patients presented with seizures of unknown etiology. For one patient, music appeared to be the trigger, and this patient tested positive for anti-GAD antibodies.
Two primary antibodies were identified in our study: anti-glutamic acid decarboxylase 65 (GAD65) and N-methyl-D-aspartate receptor (NMDA-R). Three of our patients tested positive for anti-GAD antibodies, while two tested positive for NMDA-R antibodies. The latter is typically linked to nonspecific prodromal symptoms and may eventually lead to encephalopathy or status epilepticus,8 as observed in one of our patients.
Notably, Dubey et al1 reported a different distribution of these antibodies: seven patients tested positive for NMDA-R and four for anti-GAD. However, other antibodies, such as anti-nuclear antibodies (anti-Hu, anti-Ri, and anti-Yo), and Ma-2 or PNMA family member 2 IgG, were not detected in our patients.
Furthermore, our findings indicate that the majority of seizures originated from the temporal and frontal regions, similar to the results from Divyanshu Dubey et al.’s series on autoimmune epilepsy.1
The unique electroencephalographic pattern of extreme delta brush (EDB), characterized by slow delta waves overlaid with fast activity, is most frequently associated with NMDA encephalitis. This characteristic pattern was observed in our patient who tested positive for NMDA-R antibodies.
In terms of neuroimaging, Quek et al. reported MRI findings consistent with inflammation in several patients, supporting a diagnosis of autoimmune epilepsy.9 In our patients, most MRIs yielded unremarkable results, with the exception of two cases where bitemporal lobes involvement was noted.
One noteworthy case involved the discovery of an underlying malignancy, specifically an ovarian teratoma. However, ultrasounds of the abdomen and pelvis, as well as CT scans of the chest, abdomen, and pelvis, did not reveal significant findings in the remainder of our patient group.
Upon clinical suspicion of autoimmune epilepsy, CSF analysis and comprehensive neural autoantibody screening are indicated. However, testing for selective autoantibodies is not advised, given the lack of a definitive link between any single neural antibody and seizures. Importantly, the absence of neural antibodies does not exclude a diagnosis of autoimmune epilepsy if other clinical indicators are present.9
Autoimmune epilepsy management strategies fall into two phases: acute and maintenance. Acute interventions commonly include IV methylprednisolone, IVIG, and plasmapheresis, while maintenance therapy usually involves rituximab, azathioprine, or other immunosuppressive agents.10
The IV corticosteroids are typically the first-line therapy, followed by either IVIG or plasmapheresis. Often, refractory cases necessitate the addition of a chemotherapeutic agent such as rituximab. This is supported by the findings of Lee et al11 who reported a 60% improvement rate in steroid non-responders following rituximab treatment, leading to enhanced prognosis.11 Concomitant use of immunotherapy with AEDs is clinically recommended to optimize seizure control.9
In our study, 2 patients underwent acute phase treatment involving IV methylprednisolone, IVIG, and plasmapheresis. All patients were initiated on rituximab and AEDs for maintenance therapy, resulting in favorable treatment response and improved seizure control.
Conclusion
Our case series underscores the value of early detection in autoimmune epilepsy. We recommend early diagnosis, leveraging both comprehensive clinical history and adjunctive diagnostic measures, including but not solely reliant on the antibody test. This enables timely treatment with immunotherapy, enhancing the prognosis. Furthermore, the efficacy of an initial immunotherapy regimen can substantiate the diagnosis of autoimmune epilepsy, assisting in identifying individuals who will most likely exhibit a favorable response to ongoing immunosuppressive therapy
Acknowledgement
We would like to thank Wordvice for their valuable assistance in refining the English language used in this manuscript.
- Received January 15, 2023.
- Accepted July 3, 2023.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}
{kind=link}