


Abstract
Joubert syndrome (JS) is a rare autosomal recessive disorder with cerebellar vermis hypoplasia and complex brainstem malformation. The diagnosis of cases can be difficult as the presentation can be similar to cases of cerebral palsy. We present a case of JS in an 18-month-old girl who presented to pediatric rehabilitation with a diagnosis of hypotonic cerebral palsy and abnormal eye movements. The brain MRI confirmed the typical brain malformations.
Cerebral palsy (CP) is a group of disorders of posture and movement development due to a non-progressive insult to the developing brain.1 The timing of the insult could be in the prenatal, perinatal, or postnatal period.2 Most of the clinical classifications of CP depend primarily on the tone and distribution of the motor abnormality; notwithstanding, interest in the functional classification has become popular among clinicians and therapists.3 Cerebral palsy can be classified according to the tone into spastic, dyskinetic, hypotonic, or mixed type. Spastic CP is the most common type of CP; in which, the muscle tone is increased whereas hypotonic CP is usually rare and present in children with varying degrees of reduced tone and delayed motor milestones.4 The brain MRI was found to have a strong correlation with clinical findings in most cases of CP and has helped in identifying the various etiologies.5 Our objective in presenting this particular case is to highlight the importance of thorough investigations in those labeled as hypotonic CP.
Case Report
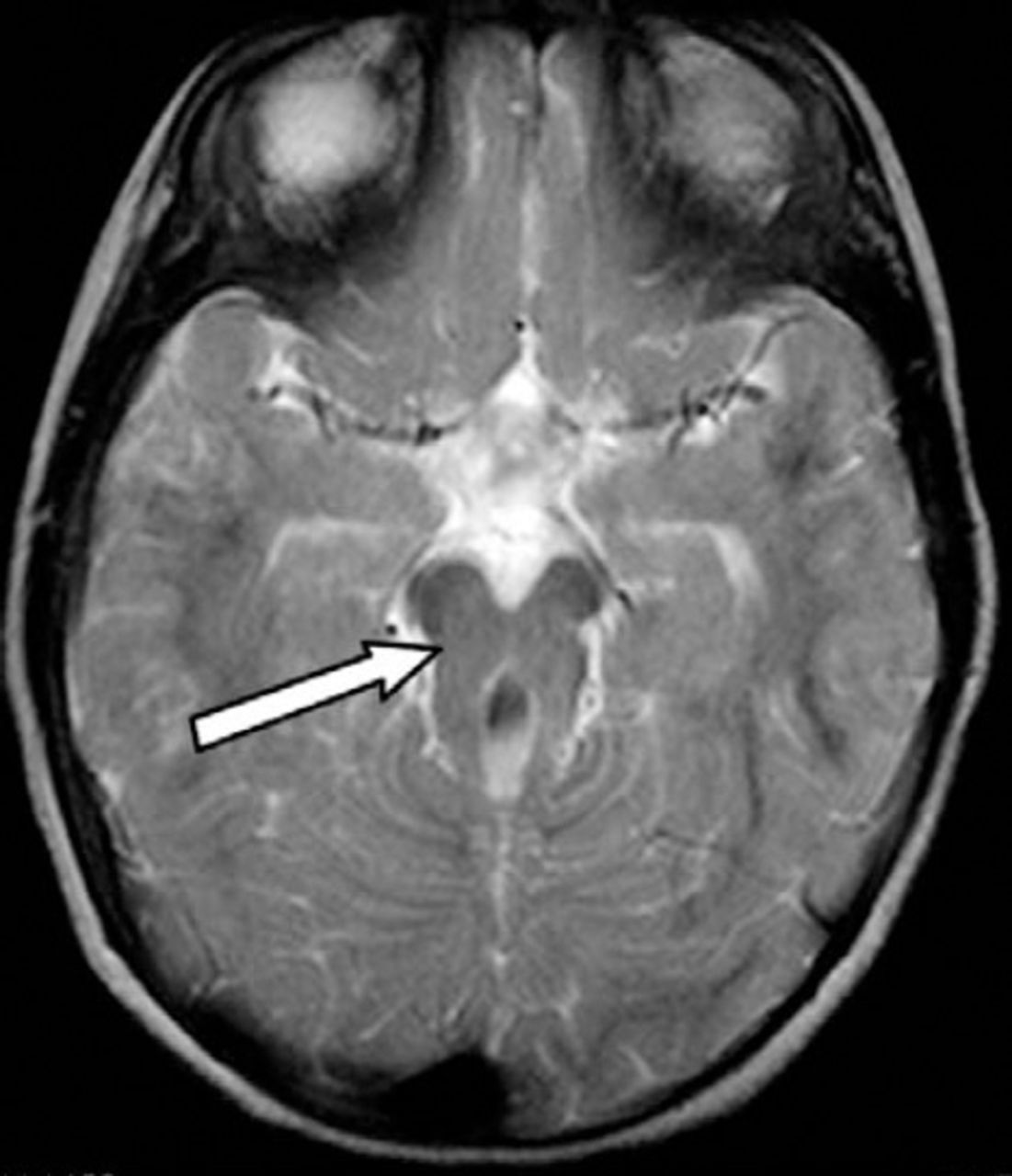
An 18-month-old girl was referred to the child development program as a case of hypotonic CP for further evaluation, multidisciplinary assessment, and therapy. She was born by emergency cesarean section due to fetal distress. Her Apgar scores were 5 and 7 at one and 5 minutes and she needed continuous positive airway pressure (CPAP) ventilation for an hour. On examination at birth, she was noted to have generalized hypotonia, but no other obvious abnormalities. At 24 hours of age, she developed tonic convulsion for which she was treated with oral phenobarbitone for one week. Brain MRI at 10 days of age showed minimal exaggeration of T2 hyperintensity of the frontal white matter bilaterally. Based on the history of fetal distress, hypotonia, and the radiological findings, a diagnosis of hypotonic CP was made. At 2 months of age, she was admitted to the hospital with labored breathing, cyanotic attacks, and abnormal rotatory eye movements. All investigations including septic screen work up, cardiac evaluation, sleep EEG, and barium study were all normal. An ophthalmological evaluation showed cortical vision impairment. It was also noted that she was hypotonic and developmentally delayed. An MRI at 6 months of age was reported with the same previous findings. On examination at 18 months of age, her weight was 13 kg (95th centile), length was 83 cm (50-75th centile), and her head circumference was 51 cm (above the 95th for age). She had marked frontal bossing and abnormal rotatory eye movements (oculomotor apraxia). Her neurologic examination showed poor head control and generalized hypotonia with lax joints but no deformity or contractures and no polydactyly. Deep tendon reflexes were present. The rest of the examination was unremarkable. Her developmental evaluation (age 18 months) based on the Schedule of Growing Skills showed delayed locomotor skills of 10 months equivalent, manipulative, visual, hearing and language skills of 8 months, interactive social skills of 10 months, self-care social skills were 12 months, and her overall cognitive skills were 8 months equivalent. The family history showed that the parents are second-degree relatives. One of the subject’s cousins has a positive history of developmental delay. In view of her hypotonia, developmental delay, large head, the history of breathing difficulties, and a positive family history, the diagnosis of Joubert syndrome was considered. A repeat brain MRI showed dilatation of the fourth ventricle with “bat wings” sign appearance (Figure 1). There was a small superior vermis, no inferior vermis was seen and a “molar tooth” appearance of the midbrain (Figure 2). Prominence of both lateral ventricles was noted with mild reduction of the thickness of the periventricular white matter and widening of the cerebral aqueduct. All previous findings were consistent with Joubert syndrome. Ultrasound examination of the abdomen revealed normal appearance of the liver, spleen, and both kidneys. Her complete blood count, renal function including blood urea, nitrogen, and serum creatinine, liver function including alanine transaminase, and aspartate aminotransferase, blood glucose, and serum electrolytes all were normal. She was referred for genetic evaluation and counseling. She was started on a rehabilitation treatment program with involvement of physiotherapy, occupational therapy, speech therapy, and orthotics services. The treatment plan focused on improving the child’s gross and fine motor skills, language, cognitive, and pre writing and social skills. On further review, 4 months after rehabilitation, she showed fair improvement in all domains of her development but continued to be delayed for her age.
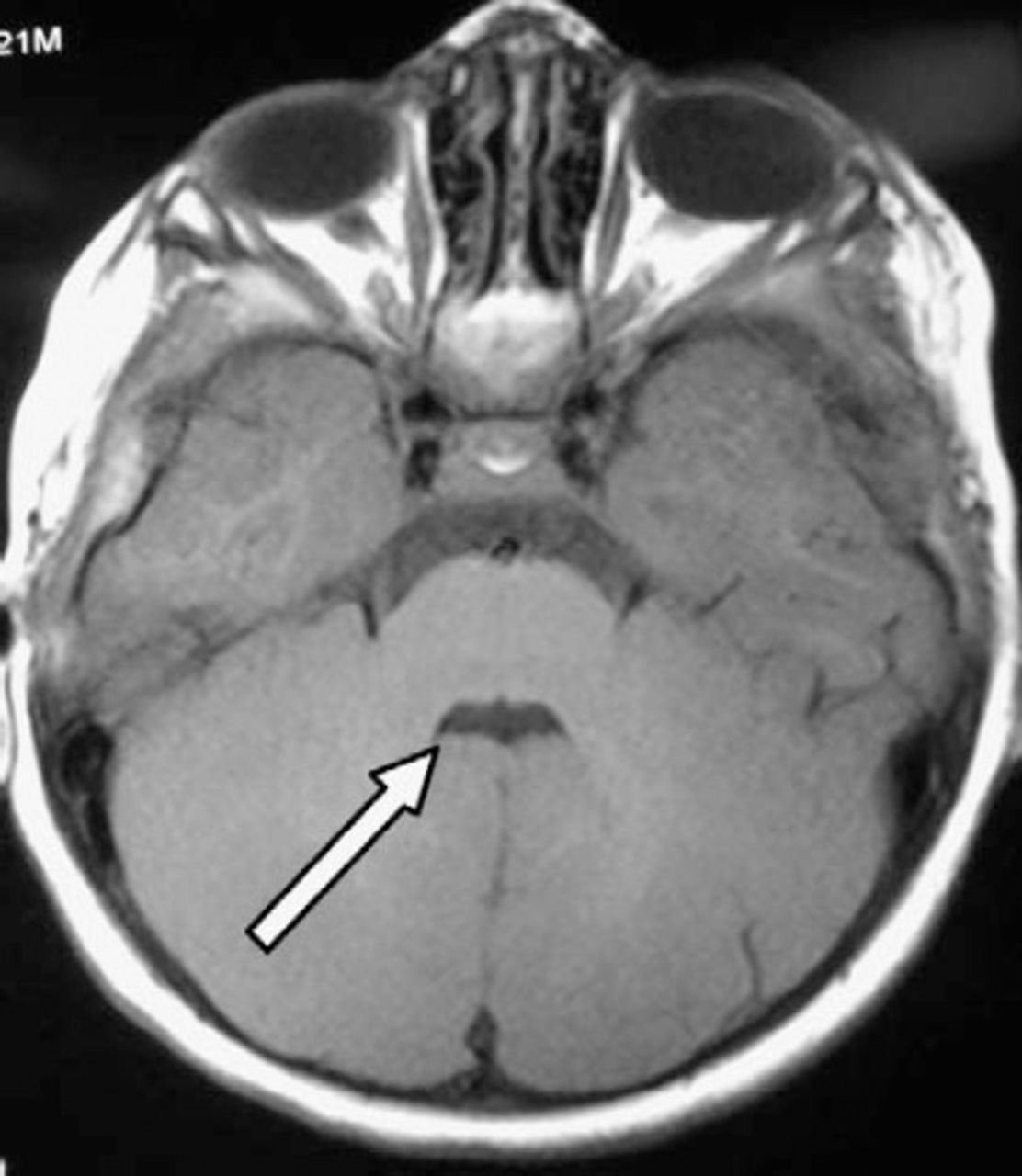
Axial T1W brain magnetic resonance sections showing “bat’s wing” appearance of the fourth ventricle and a complete midline cleft of the cerebellar hemispheres.

Axial T2W section showing “molar tooth” appearance of the midbrain due to elongation, thickening, and the horizontal orientation of the superior cerebellar peduncles and the small midbrain. Note increased depth and width of inter-pedicular distance.
Discussion
Joubert syndrome is a rare autosomal recessive genetic condition with long-term developmental, visual, and neurological consequences. It is characterized by cerebellar vermis hypoplasia, polydactyly, hypotonia, developmental delay, neonatal respiratory dysregulation, and oculomotor apraxia. Renal anomalies such as nephronophthisis or cystic renal dysplasia are common and have been reported in one-quarter of cases.6 Brain MRI imaging usually confirms the cerebellar anomalies and shows the presence of prominence of superior cerebellar peduncles, referred to as “molar tooth” sign of the midbrain-hindbrain junction.6,7
The similarity of the clinical presentation of Joubert syndrome to hypotonic CP can lead to delayed or erroneous diagnosis, as in our case. The history of fetal distress and birth asphyxia overshadowed the case and delayed the suspicion of Joubert syndrome.
Two similar cases of Joubert syndrome that were labeled hypotonic CP were previously reported; the first was a 48-year-old man who had hypotonia, learning difficulties, and oculomotor apraxia who presented with increasing tremor and gait unsteadiness.8 The second was a 25-year-old man who was diagnosed as hypotonic CP, and the diagnosis of Joubert syndrome was only suspected when he presented with repetitive cessation of breathing during sleep.9 Similar to our case, brain MRI confirmed the diagnosis in both cases.
Although Joubert syndrome is rare, it should be considered in the differential diagnosis of hypotonic CP, and when brain MRI scans are carried out, the possibility of JS, albeit rare, should also be considered. The wrong diagnosis of cases of JS can be serious as screening and identification for other associated abnormalities such as renal anomalies and breathing difficulties can be missed. In addition, unlike CP, genetic counseling is always needed in cases of JS.
- Received November 7, 2013.
- Accepted March 18, 2014.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.