


Abstract
The Dyke-Davidoff-Masson Syndrome (DDMS) results from an insult to the growing brain in utero or early infancy, which lead to loss of neurons compromising the growth of the brain. Clinical presentation includes seizures, hemiparesis, facial asymmetry, and learning disability. Radiological findings include cerebral atrophy on one side. Here, we present a case with status epilepticus who had underlying DDMS. It is a rare syndrome and uncommon cause for status epilepticus. Infections of CNS, hypoxic ischemic encephalopathy, intracranial bleed, trauma, congenital vascular malformations are the common causes of this syndrome. Diagnosis is established after clinical history, examination, and MRI. Intractable seizures can be controlled with appropriate anticonvulsants. Subsequently, these children may require physiotherapy, speech therapy, and occupational therapy in addition to the anticonvulsant medication. Outcome is better if the seizures are controlled.
The Dyke-Davidoff-Masson syndrome (DDMS) was presented for the first time in 1933 by Dyke, Davidoff, and Masson who reported a series of 9 cases.1-4 The syndrome results from an insult to the growing brain in utero, or during the early years of life.5 The insult to the developing brain results in loss of neurons compromising the growth of the brain.6 Hence, patients suffering from DDMS present with contralateral hemiparesis, seizures, mental retardation, facial asymmetry, learning disability along with radiographic findings of cerebral hemispheric atrophy on one side.2,3 It is a relatively rare syndrome, and since 1933 less than 100 cases have been reported. It may present at any age including children, adolescents, and adults.1,3 Albert Zilkha’s study comprising of 5,000 head CT scans showed 10 cases of cerebral hemiatrophy.4 Trauma, infection, stroke, ischemia, intracranial bleed, perinatal hypoxia, inflammation or congenital abnormalities including vascular anomalies are believed to be the common causes of this syndrome.2,7 These children may present with mental retardation, cerebral palsy, and epilepsy. Presentation is quite variable and they rarely present with status epilepticus (SE).8 Our objective in presenting this particular case is to highlight clinical significance of DDMS as an underlying cause of statue epilepticus.
Case Report
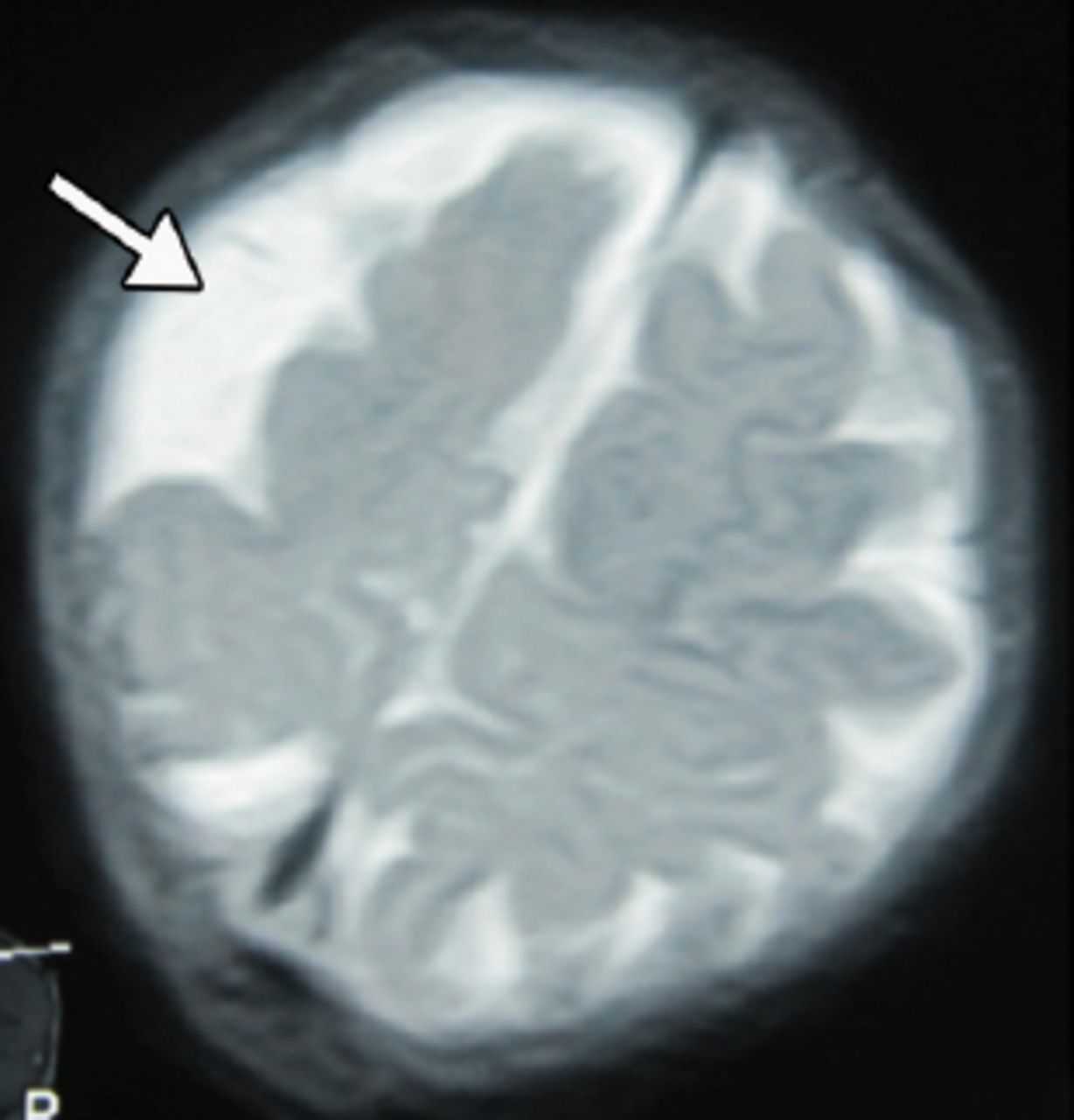
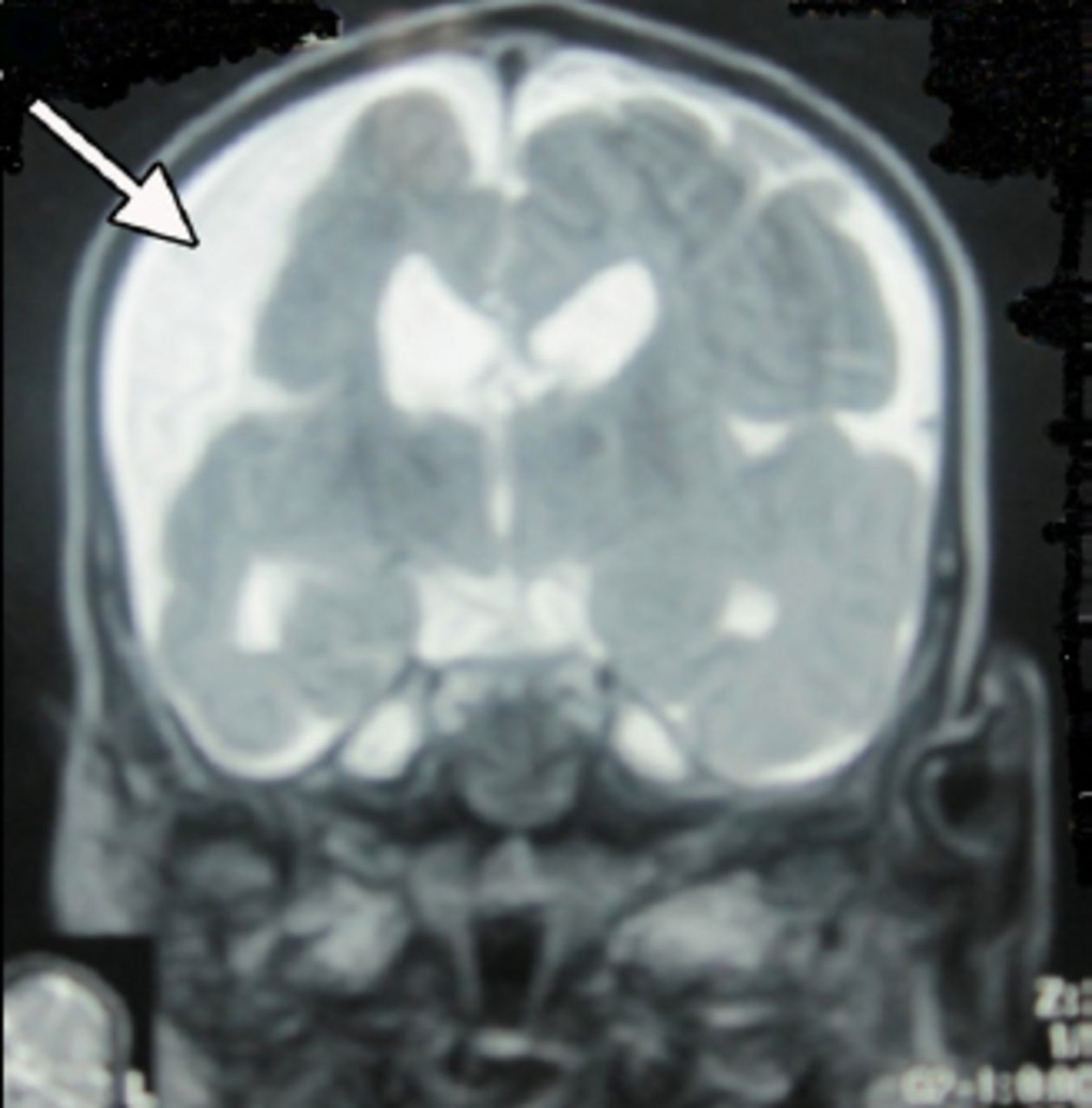
An 18-month-old boy presented with SE to the emergency room of the Children’s Hospital, Lahore, Pakistan. On clinical examination, he had left-sided hemiparesis. There was a history of meningitis at one year of age. Physical examination showed hemiparesis of the left side of the body with mild developmental delay. His septic profile and serum electrolytes were normal and the EEG showed continuous epileptogenic discharges from the right side of the brain (Figure 1). The MRI brain scans (Figures 2 & 3) revealed atrophy of the right cerebral hemisphere. Midline structures were shifted by the intact cerebral hemisphere. He was managed with supportive measures (namely, intravenous fluids, oxygen inhalation, maintenance of acid base balance and electrolytes) along with intravenous phenytoin (8 mg/kg IV), levetiracetam (40 mg/kg IV) and a continuous infusion of midazolam (8 microgram/kg/minute IV) for seizure control. After his stabilization and seizure control, he was discharged on oral phenytoin (8 mg/kg/day) and levetiracetam (40 mg/kg/day) with advice to contact the rehabilitation services and child neurology follow up clinic. His case summary was prepared, and images were taken after the informed consent from parents for medical writing and teaching purpose only.
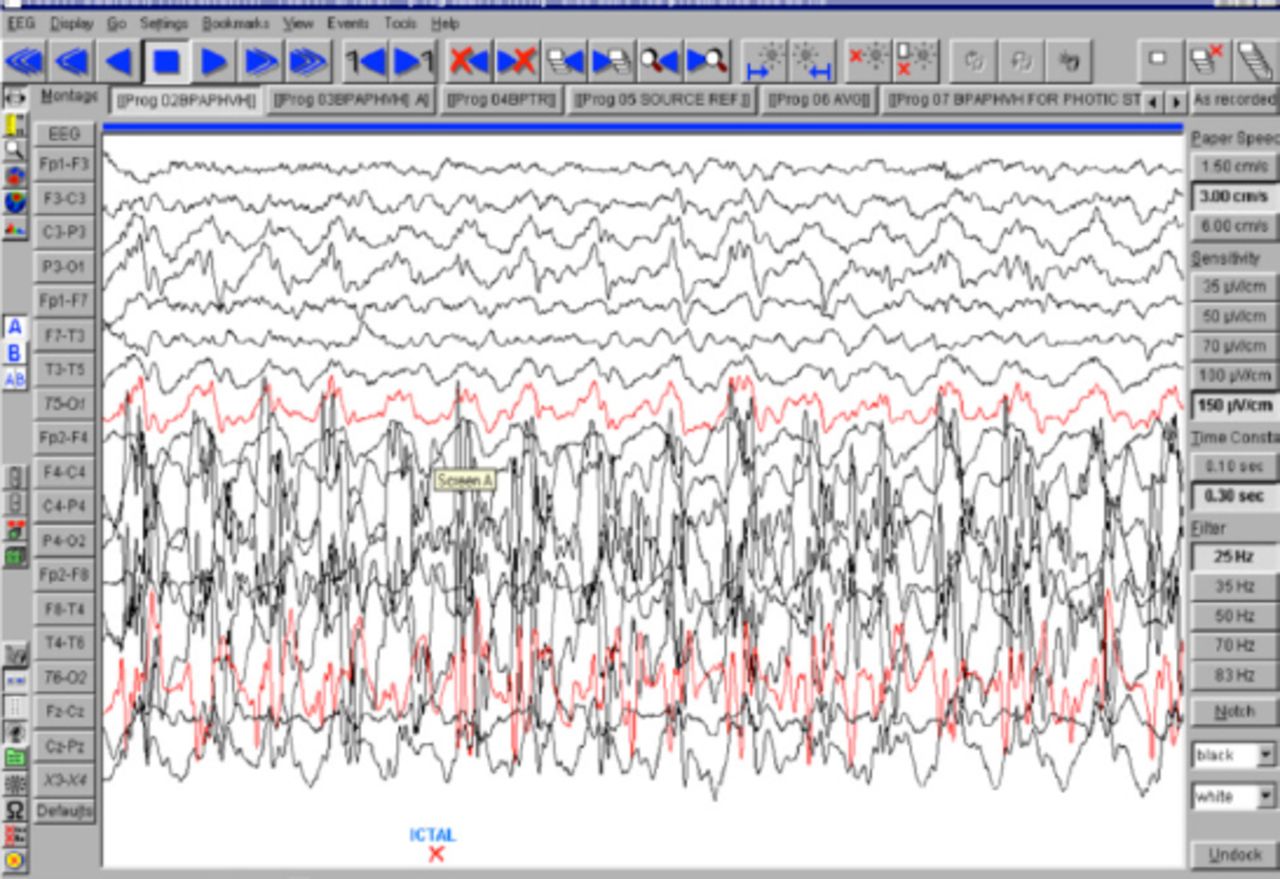
EEG showing epileptic discharges from right cerebral hemisphere
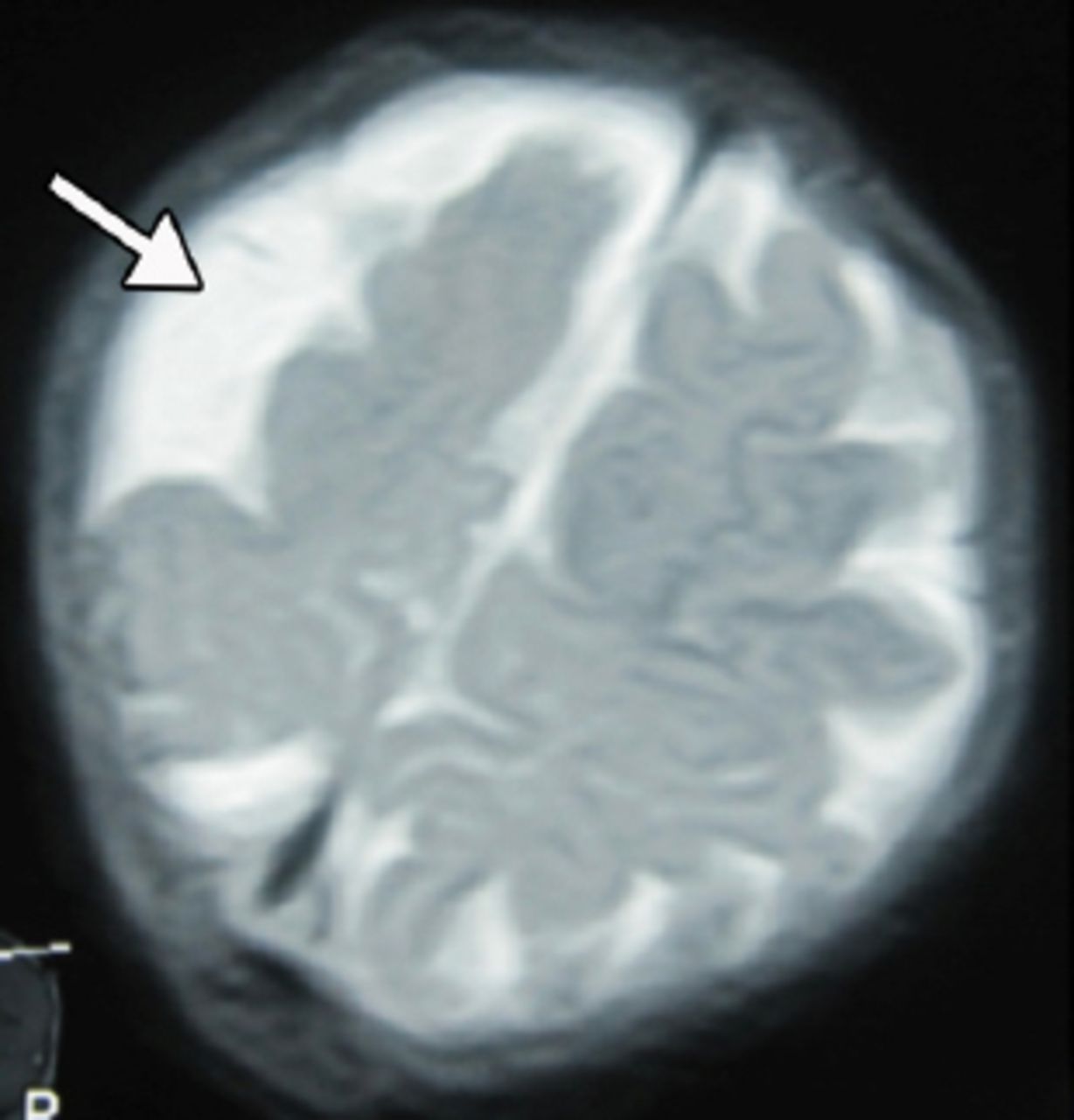
An MRI of the brain (T2 weighted images) showing right sided brain atrophy.
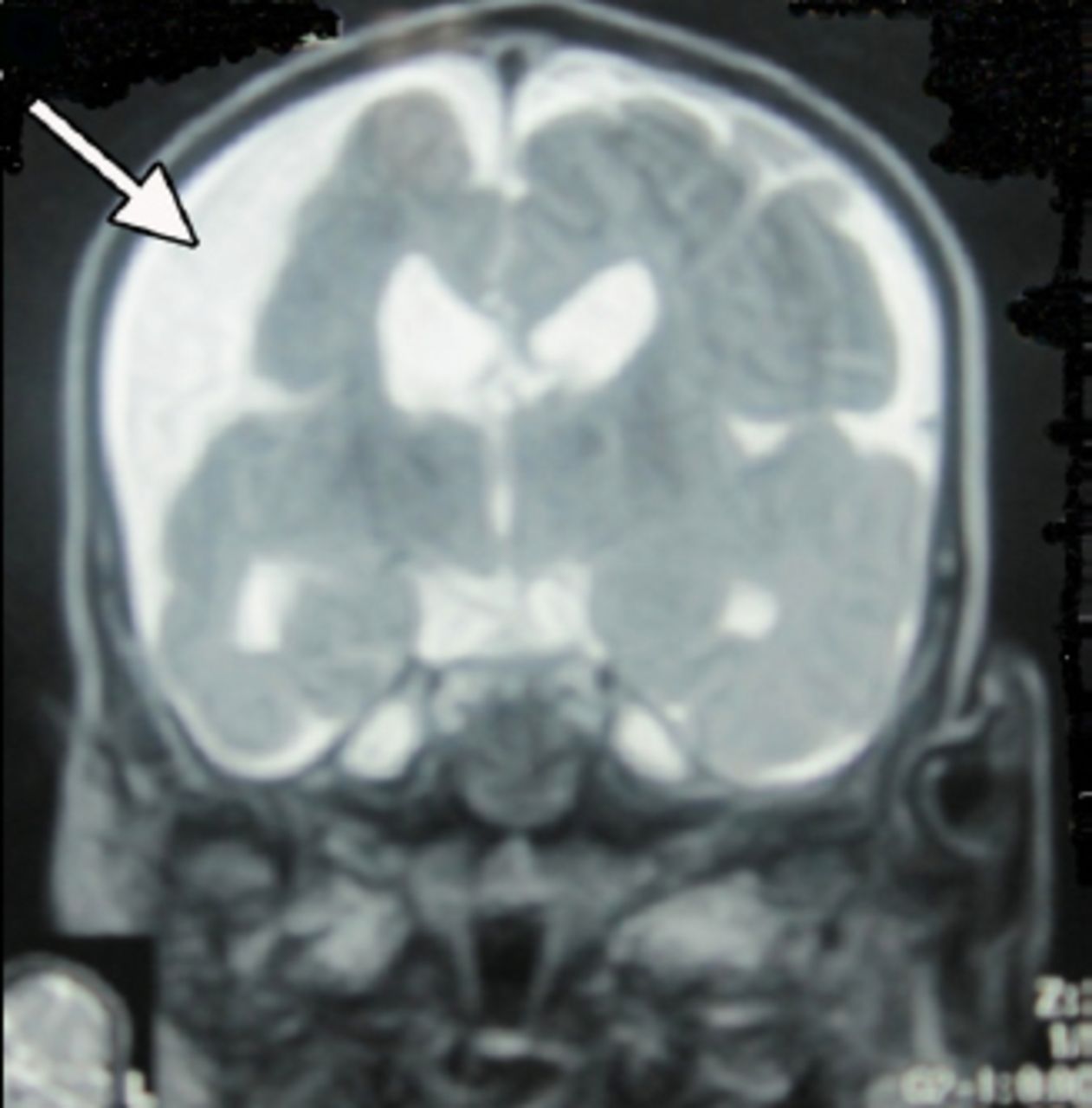
An MRI of the brain showing right sided brain atrophy.
Discussion
Dyke-Davidoff-Masson syndrome is a rare syndrome itself and presentation as SE is rarely described in the literature. Status epilepticus is one of the most common and difficult to treat neurological emergencies. The treating neurologist is in a much better position if he knows the underlying etiology. Congenital brain malformations are one of the major groups responsible for epilepsy in children.
Age of onset of symptoms largely depends on time of brain injury and specific changes may not appear until after the first decade of life.3,9 According to the degree of the brain damage, the clinical findings vary from patient to patient.3,6,8 Classically patients presents with a varying extent of facial asymmetry or paresis, sensory disturbances, hemiatrophy of body, focal or generalized uncontrolled seizures, contralateral hemiplegia, mental retardation, impaired cognition, learning and speech impairment, and psychiatric problems.3-6,8 Some reports have also described the presence of complex partial seizures with secondary generalization.3 The disease has been found to be more common in males and the left hemisphere appears to be more frequently involved.7
The disease presentations are a result of cerebral insult during gestation or early in life, and hence can be divided into 2 types, the congenital (infantile) and acquired type. In the congenital type, structural abnormalities of the cerebral vasculature seems to be the cause of this leading to cerebral injury during fetal life.1,4,6 It could also be due to some genetic defects.9
The acquired type of disease arises due to CNS injury during the perinatal period or later secondary to causes such as infections, injuries, tumors, vascular anomalies, prolonged febrile seizures, ischemia, hypoxia, and various kinds of intracranial bleeds.4-6 Perinatal hypoxia is very common in developing countries like Pakistan, due to poor obstetric care.8 A possible etiological association between DDMS and prolonged febrile seizures, schizophrenia, and middle cerebral artery stroke has been described in various case reports.8
If the disease develops within the first 2 years of life, compensatory changes, such as homolateral skull and sinus hypertrophy can result may occupy the vacuum left by the cerebral hypoplasia. The concerned side has a wide sulci replaced by gliotic cerebral tissue. In the congenital type, there is a midline shift towards the diseased side, and the sulcal prominence replacing the gliotic tissue is not present. This is what differentiates the 2 types from each other.8,9 A thorough history along with a detailed examination and appropriate imaging studies are essential to reach the diagnosis.
There are very few case reports of DDMS presenting with SE described in literature. One case is reported from Iran and described an 18-year-old girl with seizures, hemiparesis of the right side, and mental retardation who was subsequently diagnosed as a case of DDMS.10 Due to the refractory nature of the seizures, the treatment revolves around the use of one or more appropriate anticonvulsants. In the longer term, the patient needs to be managed with physiotherapy, speech therapy, and occupational therapy in addition to the medical treatment.6
Outcome is better if the onset is after 2 years of age and the seizures are controlled. For intractable seizures and hemeplegia, the management of choice is hemispherectomy.6 However, due to unavailability of hemispherectomy in many settings, the optimal way of managing the disease includes early diagnosis by appropriate imaging study, molecular genetics, medical management of seizures, and physiotherapy.11
In conclusion, DDMS is a rare clinical entity and SE is an unusual presenting complaint. The pediatric neurologist must be aware of this relatively uncommon clinical presentation of SE. The long-term prognosis is good provided the clinical entity is recognized early and managed appropriately.
- Received July 22, 2014.
- Accepted June 8, 2015.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.