


Abstract
Hemichorea is a disorder characterized by abnormal, continuous, nonrhythmic, jerky, and distal movement involving one side of the body. It may result from cerebrovascular insult to basal ganglia, or from other causes including neoplasm, infection, and non-ketotic hyperglycemia. We report the clinical, laboratory, and neuroimaging data with treatment response of a Saudi woman who has diabetes with left side hemichorea, involving the face, and upper and lower extremities, with unilateral right striatal hyperintense signal changes in T1 weighted MRI, and a hyperglycemic state of longstanding uncontrolled diabetes. Literature review suggested a syndrome with a triad of symptoms: non-ketotic hyperglycemia, hemichorea, and T1 MRI striatal hyperintensities. As the number of internationally reported cases is still modest, reporting more patients will highlight aspects pertaining to the diagnosis and treatment of this condition. We present a patient who had a sustained therapeutic result from haloperidol and clonazepam.
Hemichorea is a disorder causing abnormal, continuous, nonrhythmic, jerky, distal movement involving one side of the body. Non-ketotic hyperglycemia can cause a variety of neurological complications, such as seizures, hemianopsia, hemichorea, and coma.1 The most common cause for hemichorea is a cerebrovascular insult to the contralateral basal ganglia. Non-ketotic hyperglycemia is considered another cause for hemichorea.2 Additional causes may include hyperthyroidism, neoplasms, and infections.3 Over the past 2 decades, international reports have accumulated describing what is assumed to be a unique syndrome characterized by a triad of hemichorea, hyperglycemia, and peculiar imaging abnormalities.3 However, the number of reported cases is still modest with paucity of data from the Saudi population especially with unilateral striatal MRI changes, hemichorea, and non-ketotic hyperglycemia. The aim of this paper is to highlight aspects pertaining to the diagnosis and treatment of patients presenting with hyperglycemic hemichorea (HH). Here, we report the clinical, laboratory, and neuroimaging data of a patient with HH, discussing this topic in light of available literature on this syndrome.
Case Report
A 58-year-old Saudi diabetic female presented with left-sided involuntary movements. Unilateral movement was continuous, choreoathetotic, purposeless, non-stereotyped, and involved the face (twitching) and upper and lower extremities. It developed subacutely over 2 weeks. The movement was distressful to the patient and interfered with her activities of daily living and gait, causing recurrent falls. However, it ceased during sleep. She had poorly controlled diabetes for approximately 20 years, and was maintained on oral hypoglycemic agents. She was not on hormone replacement therapy or any other medications to which these symptoms could be attributed. There was no family history of movement disorders, and no clinical features suggestive of connective tissue diseases. Past medical history was of no relevance. Apart from the hemichorea involving the left side of the face and upper and lower extremities, her neurological examination was unremarkable; with normal higher mental function, and normal cranial and sensory nerve examination.
On admission, she was hyperglycemic with a random serum glucose of 30.3 mmol/L, (normal level <6.5 mmol/L); and her glycosylated hemoglobin was 13.5%, normal level=4-6%. Urine was negative for ketones. Expectedly, serum potassium was 5.6 mmol/L (normal level=3.5-5.0 mmol/L), and sodium was 133 mmol/L, (normal level=135-145 mmol/L), but other electrolytes were normal. Her blood count, coagulation profile, liver function tests, vitamin B12, and thyroid-stimulating hormone were all within normal. Lipid profile showed dyslipidemia (cholesterol 9.81 mmol/L, triglycerides 4.61 mmol/L, high-density lipoprotein 1.1 mmol/L, and low-density lipoprotein 6.61 mmol/L). Further workup reinforced the evidence of poor diabetic control; her ophthalmologic examination revealed moderate to severe nonproliferative diabetic retinopathy, and her protein to creatinine ratio was 2000.
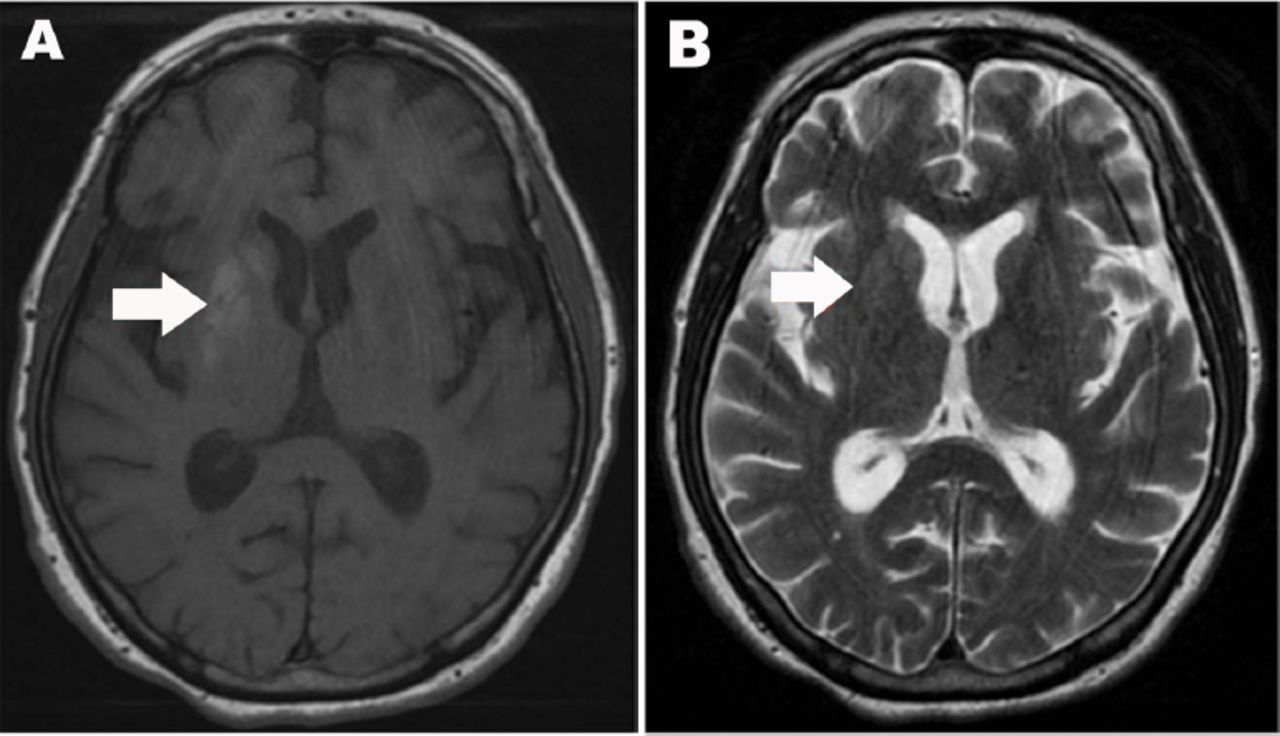
A CT scan taken on admission was normal. Non-enhanced brain MRI (Figure 1) was obtained the next day and demonstrated an area of hyperintensity on T1 weighted images; involving the left caudate, putamen, and internal globus pallidus. There was no restriction on diffusion-weighted images (DWI) and the apparent diffusion coefficient (ADC) map was normal. For glycemic control, she was started on an insulin regimen (bedtime insulin glargine and preprandial insulin aspart) with sitagliptin.

Axial MRI of the brain A) Axial T1-weighted MRI of the brain showing right striatal increased signal intensity including right putamen, globus pallidus, and head of caudate. B) Axial T2-weighted MRI demonstrate normal signal intensity.
Initially she was treated with lorazepam (2-6 mg/day) to control her HH. While lorazepam improved the quality of her sleep, it had no effect on the chorea in her waking hours. Valproic acid (400 mg/day) had no clear effect on chorea and was discontinued due to intolerance. She was then given haloperidol (gradually titrated from 1 mg/day to 8 mg/day) and clonazepam (titrated from 1.5 mg/day to 6 mg/day). She showed clinical improvement after a few days with improvement in her daily activities and gait. Initially, there was more improvement in her lower extremities than upper extremities and the face over a period of 2 weeks. However, she achieved a satisfactory glycemic control, with a fasting serum glucose ranging from 6 to 7.8 mmol/L. With the initiation of neuroleptics and the glycemic control, the hemichorea steadily improved. She was discharged and followed up at an outpatient clinic. Three months later, she was in stable condition and had no recurrence of chorea.
Discussion
Several studies2,3,5,6 investigated the triad of hemichorea or hemichorea/hemiballism, non-ketotic hyperglycemia, and striatal hyperintensity, and have suggested a new syndrome.3 Patients suffering from HH share common clinicoradiologic features. In examining this Saudi patient, we regarded this as a case of HH due to the unilateral choreoathetotic movement, hyperglycemia on presentation with poor glycemic control, and absent urine ketones, contralateral TI hyperintensity in the basal ganglia with no restriction on the ADC map, and the absence of other metabolic derangements.
Elderly patients are more susceptible to HH. The average age of onset ranges from 64 to 82 years.3 Earlier case reports of HH suggested prevalence in females,4 but literature reviews indicate equal male to female incidence,3,5 which emphasizes the importance of accumulating case reports to better understand epidemiological characteristics. The patient in this study falls into the typical gender and age group for HH. Interestingly, there is a possible racial predisposition to HH. Lin et al3 estimates that 86% of the reported cases of HH were Asians. Abnormal movements usually involve one side of the body, but a few patients with bilateral movements were reported.6,7 Some patients were newly diagnosed diabetics, and their HH was the first manifestation of the disorder.3,4 Others, as in our patient, had longstanding diabetes.3,5,8,9
Hyperglycemic hemichorea has a good prognosis and hemichorea rapidly ceases over a few days to weeks, after restoring glycemic levels with insulin only.3,5,8 There is enough evidence to speculate that radiological changes are also reversible. Patients who had follow-up imaging studies showed resolution of changes on CT within 2-3 months and on MRI within 6-11 months.3 However, a minority of cases had a different course. Some patients did not improve on insulin alone, and required either short or long courses of additional pharmacologic therapies. Ahlskog et al,4 and others reported cases with hyperglycemic dyskinesia that persisted as long as the patients were followed, 6 months up to 5 years, with patients showing no or slight improvement. These persisted despite the normalization of blood glucose and the use of pharmacologic agents.4,7 Recurrence of symptoms after drug discontinuation was also reported in the literature.7 A few patients responded very poorly to all therapeutic measures and eventually died of secondary medical complications.6,10
Several neuroleptic and anti-epileptic agents were cited in the literature to control HH. Haloperidol (1-15 mg/day) frequently showed good efficacy in managing HH,2-4,9 but dose escalation in refractory patients can be limited by side effects, such as tardive dyskinesia or hemiparkinsonism.7 Moreover, risperidone (3 mg/day) may be effective.4 The response to valproic acid was inconsistent. Trials of lorazepam,10 chlorpromazine (12.5 mg/day), clonazepam (5 mg/day), reserpine (1.5 mg/day),8 and tetrabenazine (50 mg/day)2 lacked sufficient evidence of their effectiveness. Trihexyphenidyl (6 mg/day), olanzapine, perphenazine, and biperiden were of no benefit.7,9 Our experience supports the effectiveness of haloperidol, and the potential effectiveness of clonazepam while discouraging the use of lorazepam.
In addition to the distinctive clinical features of HH, it has characteristic findings on brain imaging. Brain CT may show subtle hyperattenuation in the contralateral striatum. Brain MRI gives a high-signal intensity on T1 images and normal- to low-signal intensity on T2 images.4 These observations align with the imaging findings in our patient. Gradient echo images, DWI, and ADC maps are usually normal,4,5 susceptibility weighted imaging (SWI) is sensitive to blood products similar to blood oxygen level-dependent imaging. In cases of hypo perfusion of brain tissue, local vasodilation occurs in the ischemic tissues, which leads to increased extraction of oxygen and increased amount of deoxyhemoglobin in venous blood in that region. As deoxyhemoglobin is paramagnetic, it shows prominent hypointense signal on SWI, and can be detected as hypointense venous vessels in the affected area.2
Additional insights are gained by more sophisticated imagining modalities. Magnetic resonance spectroscopy shows a decreased N-acetyl aspartate peak, which indicates a neuronal dysfunction or loss resulting from metabolic failure or hypoperfusion.6 Positron emission tomography demonstrates reduced fluorodeoxyglucose (FDG) uptake in the corresponding basal ganglia, especially when performed later in the course of the illness,8 while showing increased FDG metabolism when performed earlier.9 Similarly, single photon emission computed tomography (SPECT) studies show either increased8 or decreased cerebral perfusion of the affected stratum. This discrepancy, as noted by Hsu et al,8 may be secondary to changes of cerebral vascular autoregulation or to the different stages of the pathological process at which the SPECT studies were carried out.
The underlying pathophysiological mechanism for HH is still uncertain. Several sound hypotheses have been proposed to explain the neuroimaging findings of such cases: mineralization, hyperosmolar insult, petechial hemorrhages, gliosis, transient ischemia, manganese accumulation, and Gamma-amino butyric acid (GABA) transmission alteration. The reversibility of neuroimaging findings on follow-up studies rules out calcification as a possible cause. Gamma-amino butyric acid deficiency in the basal ganglia as a result of its consumption as an energy substrate during hyperglycemia has been suggested.9 This fails to explain the unilaterality of symptoms and its persistence in some patients, as in this case study, beyond the hyperglycemic episode.7 Several authors6,10 suggested that striatal petechial hemorrhage is the etiologic mechanism for HH and its T1 hyperintensity. Only one histopathological autopsy report6 mentions hemosiderin deposits in a patient with HH, whereas another report10 describes microhemorrhages. These microhemorrhages supposedly result from hyperglycemia-induced blood brain barrier dysfunction. However, several observations are difficult to reconcile with this hypothesis: lesions frequently respected anatomic borders, T2 images are not hyperintense, and imaging evolution over time is not typical for bleeds.7
Histopathological studies of cases with HH have heterogenous results, demonstrating infarcts, microhemorrhages, gliosis, and mineralization.10 Nevertheless, they all have gemistocytic gliosis as a common findings. Fujioka et al,5 experimentally reproduced similar striatal T1 hyperintensities and T2 hypointensities in rats by means of brief focal ischemia, which showed matching histological finding to cases of HH: selective neuronal death and astrocytosis rather than infarcts or bleeds.
While the definite mechanism is still debated, it is fair to assume that a combination of the aforementioned processes is responsible for HH.7 Due to microangiopathy, diabetics, especially poorly controlled diabetics, are predisposed to vascular ischemia, especially during a hyperglycemic crisis. A partial ischemic injury may cause selective neuronal death and gemistocytosis, interrupting striatal GABAergic transmission, disinhibiting the thalamus, and leading to abnormally excessive movement.
In conclusion, diabetics are susceptible to HH, which is associated with T1 MRI hyperintensity. Clinicians should recognize this unique syndrome early as it is treatable with normalization of blood glucose. For patients who do not improve on insulin alone, we see a good therapeutic role for haloperidol, and a potential role for clonazepam. Further research is needed to elucidate the underlying pathophysiology of HH.
Footnotes
Disclosure
Authors have no conflict of interest, and the work was not supported or funded by any drug company.
- Received August 19, 2015.
- Accepted November 4, 2015.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.