


Abstract
Brainstem encephalitis (BE) is a rare, severe, and potentially life-threatening inflammation of the central nervous system. Brainstem encephalitis has multiple etiologies, which vary in treatment and outcomes. The current literature is generally focused on the infectious causes of BE, while little is known about the other entities, including cases with inconclusive diagnoses. Additionally, the outcomes of BE are not well documented. We present a case of an 18-year-old male who presented with progressive symptoms of brainstem involvement. His clinical investigations, including cerebrospinal fluid (CSF) analysis, were normal; magnetic resonance imaging (MRI) of the brain showed an enhancing medullary lesion, while tissue biopsy yielded no specific diagnosis. Multiple empirical treatments to address possible autoimmune and infectious processes were started with no significant improvement. He continued to deteriorate over a period of 12 weeks. Thereafter, following intensive supportive and rehabilitative care, he started to show slow signs of improvement.
Brainstem encephalitis (BE) is a relatively rare, severe, and rapidly progressive inflammation of the brainstem.1 This challenging entity is largely associated with infectious and autoimmune causes. Additionally, some cases do not fit within these categories, meaning that making a diagnosis can be quite challenging and prolonged. The clinical course, prognosis, and treatment outcomes of BE vary accordingly.2 The current literature on this group of disorders mainly consists of case reports documenting instances of BE with infectious origins. The other entities – specifically, those cases where all adjuvant investigations are inconclusive – are rarely described. The clinical presentation of BE includes ataxia, cranial neuropathies, altered levels of consciousness, and long tract involvement. Moreover, a few distinct features can be seen in some variants of BE. For example, fever, headaches, and chills raise the suspicion of an underlying infectious event. Further, a previous history of an upper respiratory tract infection is usually associated with an autoimmune or inflammatory event.1,3 In the current report, we present a case of severe BE that progressed rapidly, showing normal clinical investigations apart from an enhancing lesion on brain magnetic resonance imaging (MRI). The patient made a significant recovery following extensive supportive care. We also uniquely present the histopathological findings of such a challenging entity.
Case Report
Patient information
An 18-year-old, previously healthy male presented with a history of diplopia and right-sided facial weakness that developed 3 weeks prior to presentation. Two days later, he developed symptoms of oropharyngeal dysphagia, slurred speech, and decreased hearing on the right side. One week later, he developed unsteady gait with a tendency to fall on his right side, which was accompanied by decreased temperature sensations in the left upper and lower limbs. His symptoms were preceded by an upper respiratory tract infection that was treated with antibiotics. His family history was unremarkable. Figure 1 summarizes the timeline of the clinical event.
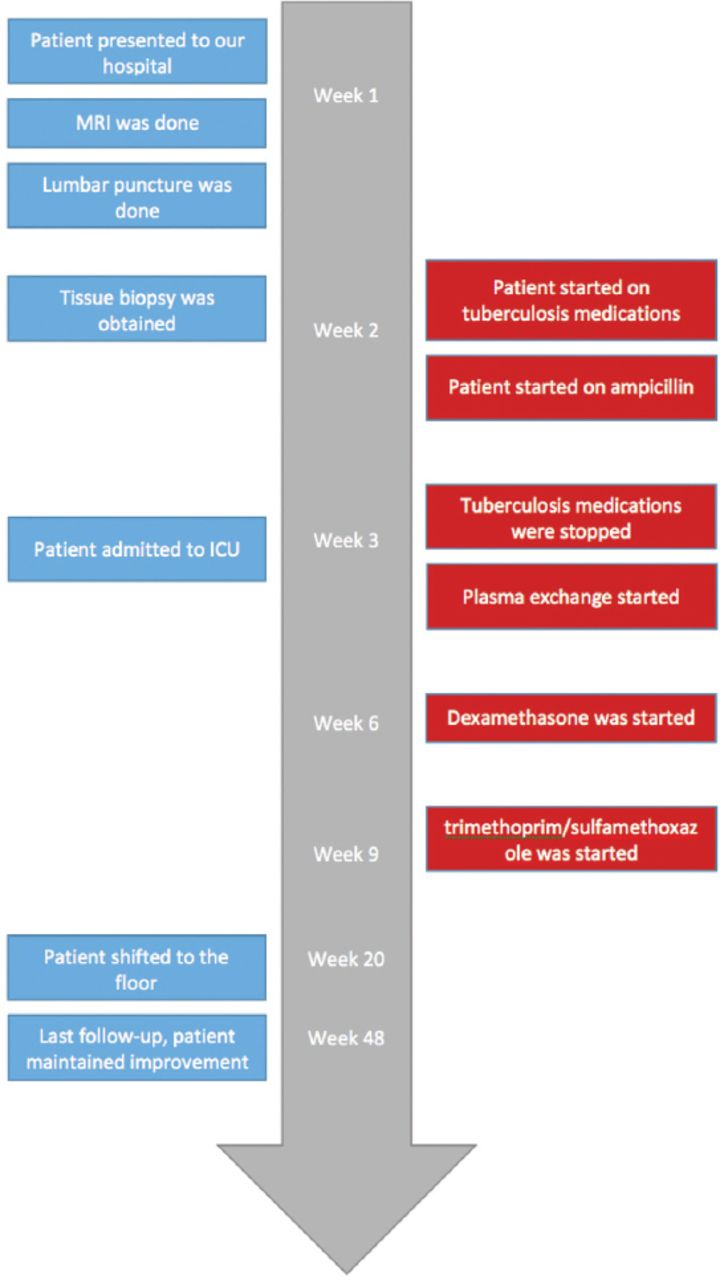
Timeline figure of the clinical event.
Clinical findings
On physical examination, he was vitally stable, conscious, alert, and oriented to place, person, and time. Cranial nerve examination determined right-eye ptosis, the primary gaze of the right eye was in an in-and-down position, while the primary gaze of the left eye was in an out position. An extraocular muscle assessment showed limitations in right-eye abduction and left-eye adduction. Furthermore, there was a horizontal, rapid, jerky nystagmus in the left gaze, an absent gag reflex, and grade V House-Brackmann right facial palsy. Motor examination showed a power of 4 out of 5 in the right upper and lower limbs. Pain and temperature sensations were decreased in the left upper and lower limbs. Cerebellar and gait examination showed impaired finger-to-nose and heel-to-toe test results on the right side, with an inability to walk due to imbalance.
Diagnostic assessmen
Routine bloodwork such as complete blood count, coagulation profile, renal profile, and liver profile were within normal limits. Other investigations that were performed to exclude infectious and autoimmune causes did not show significant results. Table 1 summarizes the laboratory investigations that were conducted with the patient described herein.
Laboratory findings of the patient presented.
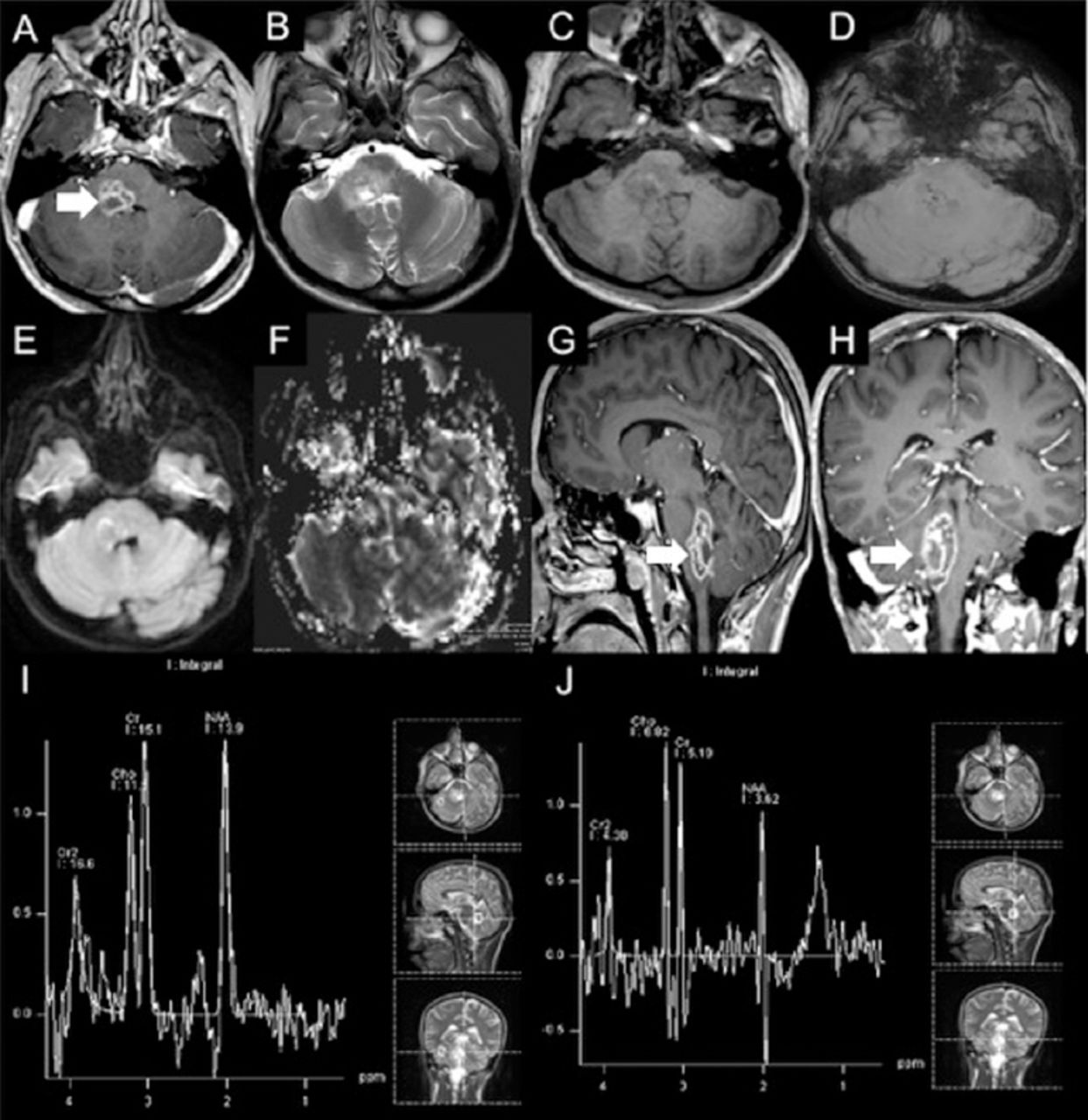
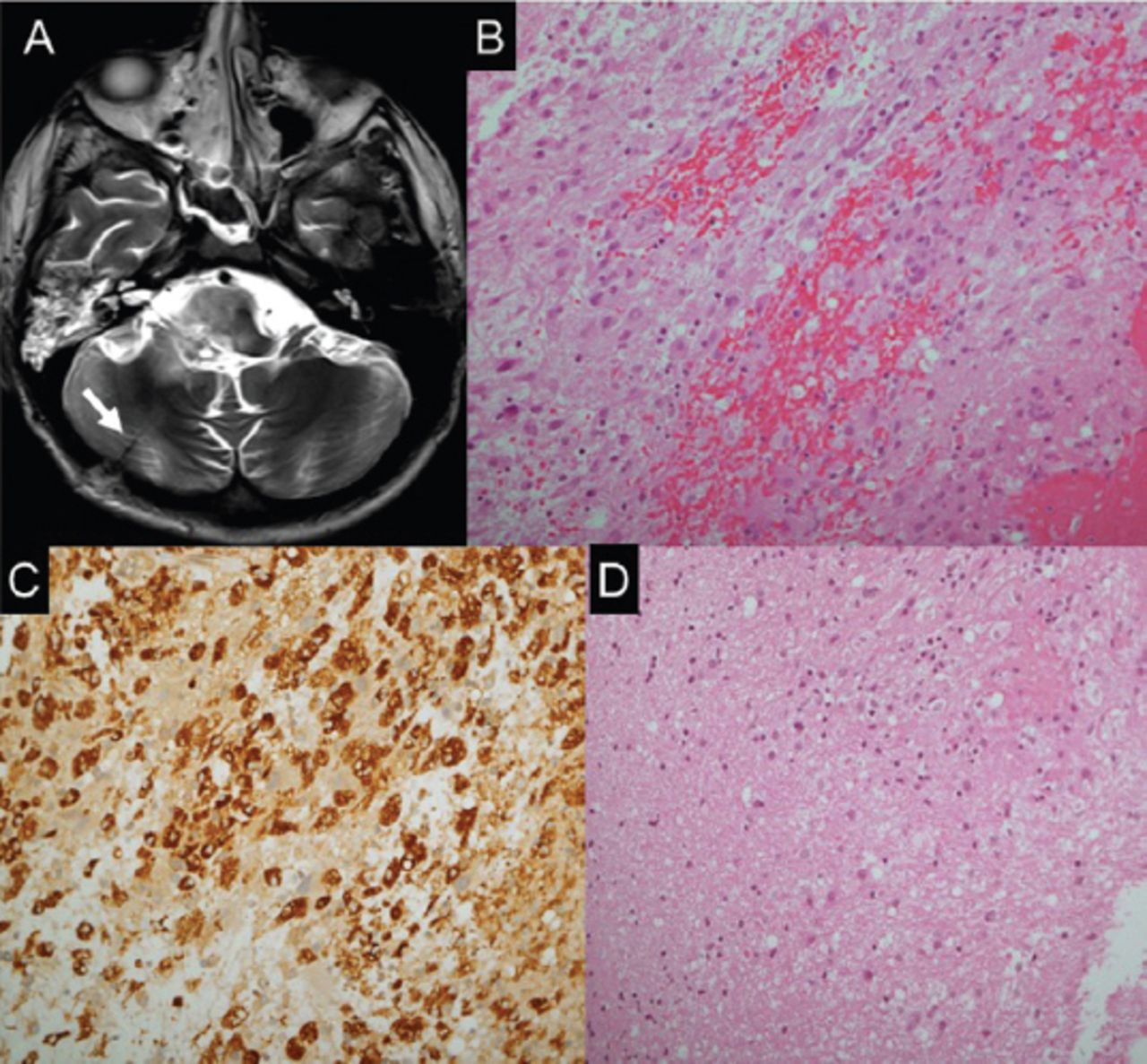
Lumbar puncture showed normal results. Brain MRI showed an enhancing focal lesion on the right side of the medulla oblongata, which was posteriorly located and featured an extension into the middle cerebellar peduncle and posterior pons (Figure 2). Magnetic resonance (MR) spectroscopy showed decreased N-acetylaspartate (NAA) and increased choline peaks. There was also an increased lipid-lactate peak (Figure 2). Stereotactic brain biopsy was carried out to exclude neoplastic and infectious causes. The findings included areas of gliosis and foci of chronic inflammatory cells, in keeping with encephalitis (Figure 3).
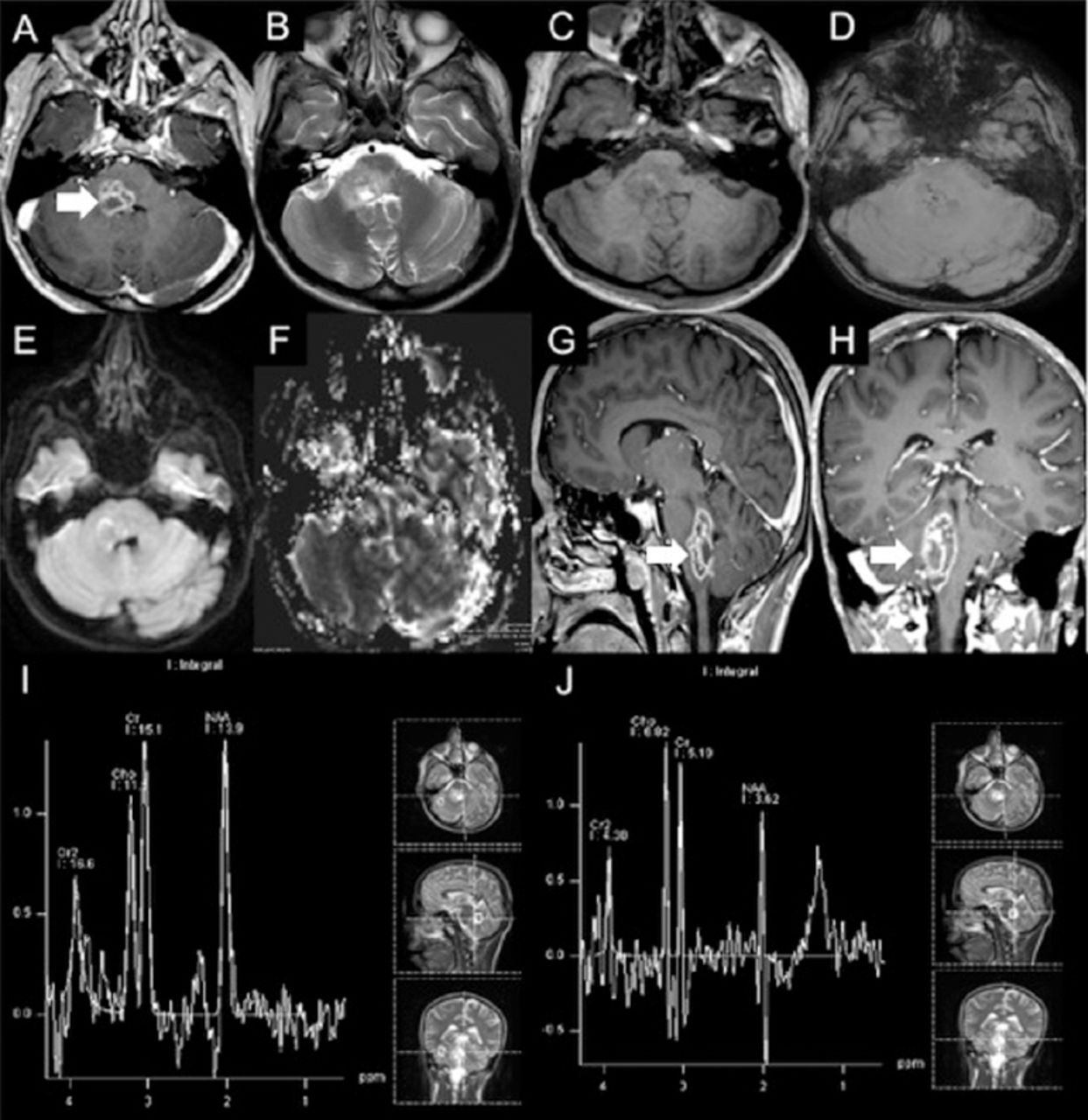
Initial brain MRI showing A, G & H) focal lesion in the right side of medulla oblongata posteriorly with extension into middle cerebellar peduncle and posterior pons. Post contrast images show multiple ring-like enhancing areas are around the central hemorrhagic focus. D) Susceptibility weighted image (SWI) shows multiple foci of dark signal indicating hemorrhagic foci. E) Diffusion weighted image (DWI) shows no restricted diffusion in central non enhancing area to suggest any purulent material. F) Central hemmohagic areas give relatively increased rCBV values. The surrounding FLAIR hyperintensity show slightly lower rCBV values compared to normal appearing cerebral white matter. I) Single voxel MR spectroscopy at TE of 135 with region of interest in normal appearing tissue around the lesion shows nearly normal NAA, choline and J) creatine peaks and at TE of 135 with region of interest in the lesion shows decreased NAA and increased choline peaks. There is also increased lipid-lactate peak around 0.9 to 1.5 ppm.
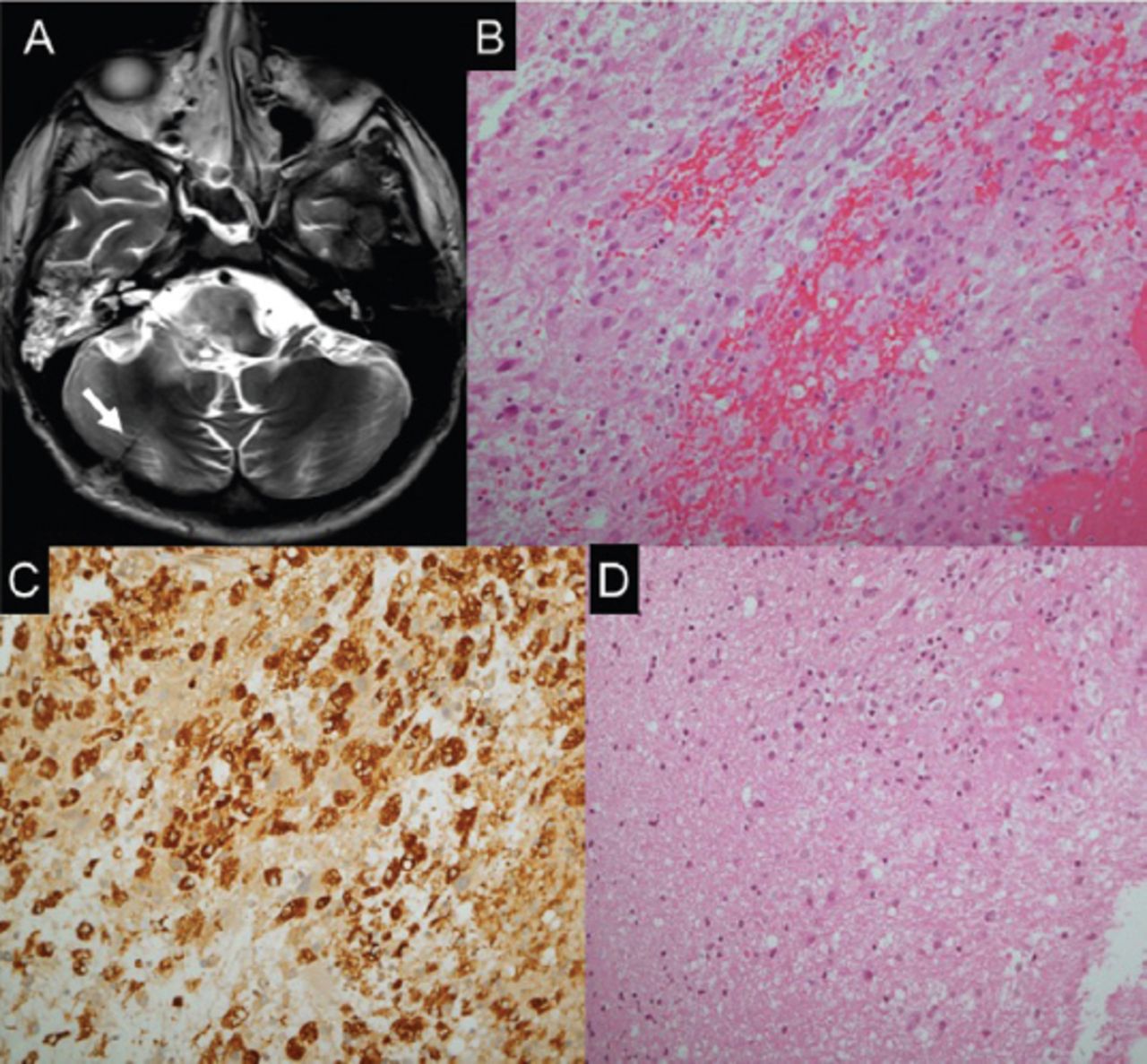
Histopathological findings. A) Axial T2 weighted brain MRI showing post biopsy changes at the right cerebellar hemisphere. B) The biopsies exhibit fragments with cellular areas, formed by mixture of chronic inflammatory cells, with dominance of histiocytes, in addition to lymphocytes, plasma cells and glial cells. C) A major component of the cellular infiltrate reacts to CD 68, confirming histiocytic nature. D) In areas, the CNS tissue sampled exhibit gliosis and foci of chronic inflammation, in keeping with encephalitis.
Therapeutic intervention
During the patient’s hospital stay, he received multiple empirical trials of treatment for a possible infectious and autoimmune event. For infectious coverage, the patient received anti-tuberculosis medications (isoniazid, rifampin, ethambutol, and pyrazinamide) for one week, which were stopped after excluding tuberculosis. The patient also received ampicillin for 8 weeks and trimethoprim/sulfamethoxazole for 10 weeks. For autoimmune coverage, he received 5 sessions of plasma exchange, as well as 2 administrations of dexamethasone pulse therapy; however, these treatments did not yield any initial benefits and he continued to deteriorate clinically and radiologically, warranting intubation and admission to the critical care unit where he received supportive care.
Follow-up and outcomes
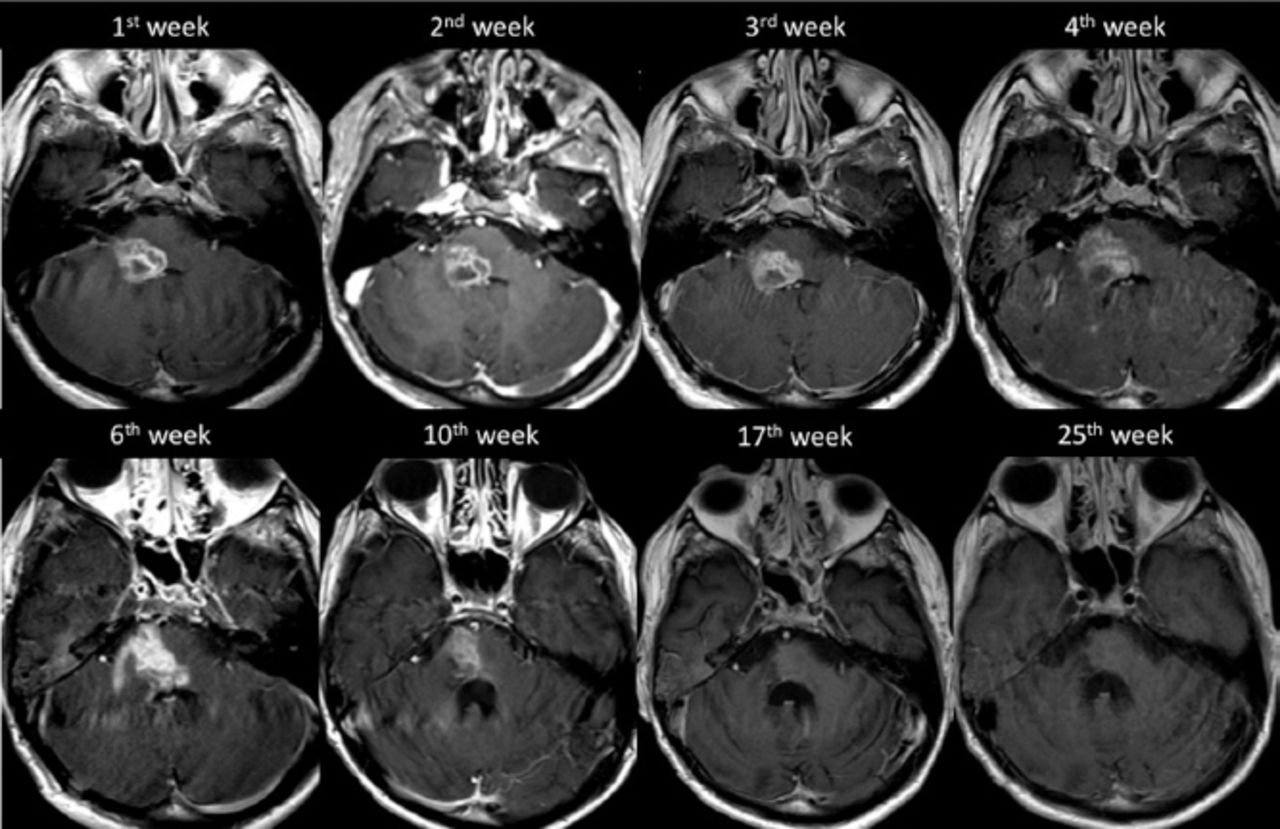
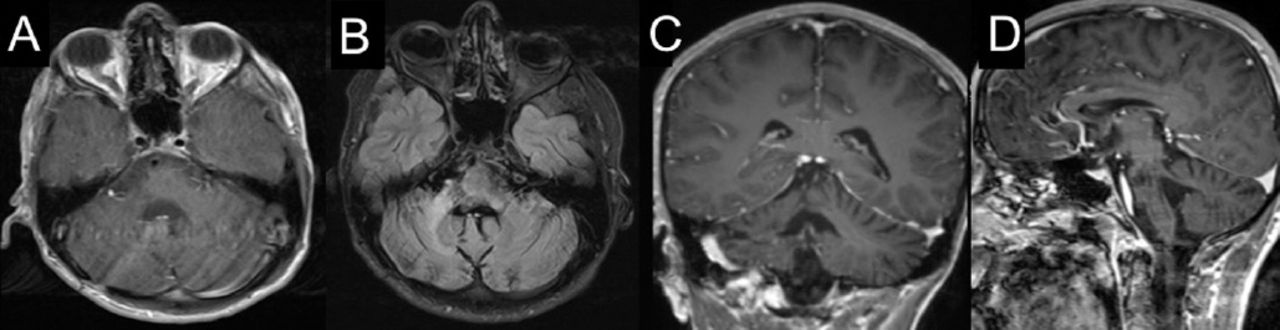
After 12 weeks from the time of clinical presentation, he started to show slow signs of improvement, both clinically and radiologically, suggesting a resolution of the inflammatory process (Figure 4). In our last follow up, one year after admission – and following intensive supportive and rehabilitative care – he maintained his improvements and could walk independently. Additionally, a follow-up MRI carried out at the 57th week of presentation showed regression in the size of the lesion (Figure 5).
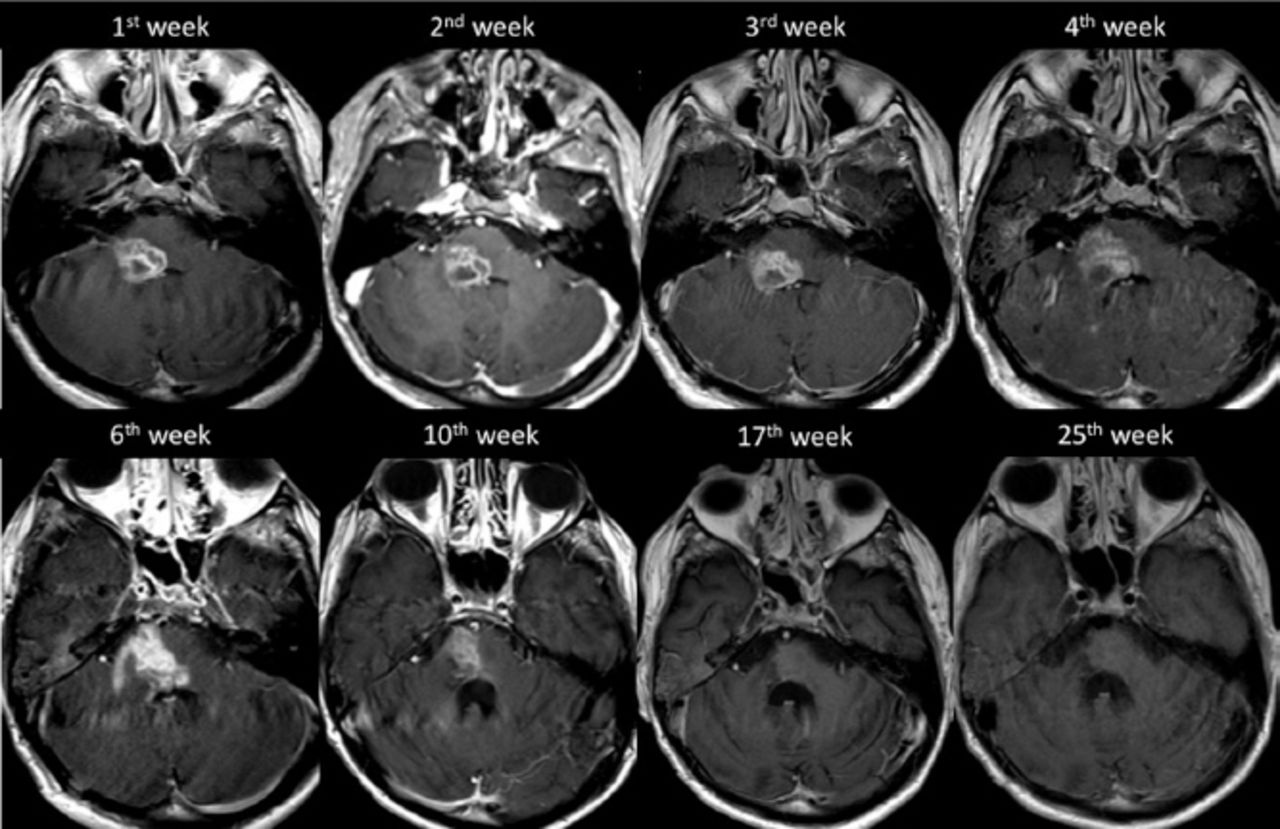
The progression of the disease demonstrated on axial T1 weighted post contrast imaging.
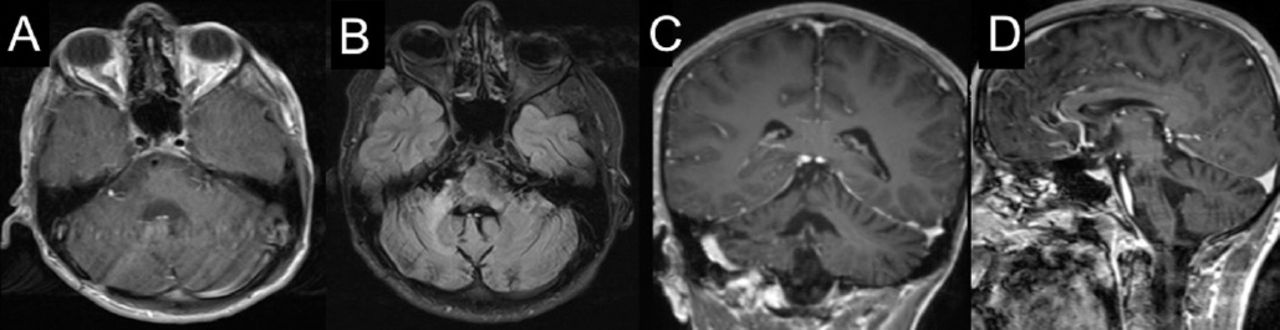
Follow-up brain MRI at 57th week of presentation. (A) axial, (C) coronal, (D) sagittal T1 weighted post contrast imaging and (B) axial flair imaging show regression in the size of the lesion at the right aspect of the pons and right middle cerebellar peduncle with a mild volume loss of the right side of pons.
Discussion
Encephalitis is described as a rapidly progressive inflammation of the brain parenchyma with an estimated incidence of around 5-10 per 100,000 inhabitants per year.4 Encephalitis involving the brainstem is relatively uncommon, with only a few cases reported in the literature. Moreover, BE tends to behave aggressively, showing debilitating neurological sequelae.3 The clinical course of BE can progress rapidly, leading to respiratory failure and warranting intubation, as was observed in our case.
The proposed etiologies of BE include infectious and autoimmune causes.1,3 The existing knowledge of BE, or encephalitis in general, is usually directed toward infectious entities. This can be explained by the existence of diagnostic and management guidelines for infectious causes, while such standards are generally lacking for other entities.4 Over the last decade, more attention has been paid toward the other possible causes of BE. This was documented in published series of BE that discussed various etiologies. In 2 reported series by 2 different authors, it was found that among 97 patients of the first study and 81 patients of the second study, most cases were attributed to an autoimmune or inflammatory process rather than an infectious process.2,3 More importantly, a significant number of cases did not fit the categories described; 31 and 23 cases were of an unknown etiology.3,2 This was also noted in a study of general encephalitis where 50% of cases were of an unknown cause.5
The diagnosis of BE can be challenging in most cases. Nonetheless, adjuvant serological markers, cerebrospinal fluid (CSF) examination, radiological evaluation, and histopathological confirmation of the diagnosis are of paramount importance. The utility of serological markers in cases of BE is to detect those antibodies that have been described to be of an autoimmune nature. Lumbar puncture for CSF chemistries (which include white blood cell counts, as well as protein and glucose levels) and the need to obtain cultures are essential to exclude infection. The role of brain imaging, computed tomography (CT), and MRI is helpful to narrow the differential diagnosis. As was evident in our case, brain MRI was not sufficient for differentiating between neoplastic and other lesions; therefore, tissue biopsy was indicated. The utility of tissue biopsy in BE cases was only reported in one previous study.2 In their series of 81 BE patients, biopsy was obtained in 14 cases. Biopsy pointed to a specific diagnosis in 50% of cases; there were 4 cases with demyelination, 2 cases with neurosarcoidosis, and one case with chronic lymphocytic inflammation and pontine perivascular enhancement responsive to steroids (CLIPPERS). Similar to our case, the remainder of their biopsied cases (7 patients) showed no specific diagnosis.
The treatment of BE is highly variable. If an infectious cause is suspected, specific antimicrobial or empirical treatments are warranted. Ampicillin is usually the drug of choice in suspected cases of listeria; alternatives include trimethoprim-sulfamethoxazole, vancomycin, and linezolid. Cases of viral BE, specifically those caused by herpes, can be treated effectively with acyclovir. While the involvement of tuberculosis in BE is rarely encountered, the proper administration of anti-tuberculosis medications is essential.1 The treatment of BE attributable to an autoimmune cause includes steroid therapy, plasma exchange, and intravenous immunoglobulin. When no etiology is established for BE, treating it as an autoimmune condition can be effective.1,2 However, in clinical practice, treatment of BE is usually initiated empirically even without clinical or laboratory confirmation, as was observed in our case.
The outcomes of BE are rarely reported. In one study, it was found that CSF parameters (glucose and protein in particular) played a role in predicting outcomes. Authors have observed that patients with high CSF protein and glucose levels exhibited poor outcomes.2 Our patient showed good outcomes, possibly explained by their findings, as this patient had nearly normal CSF protein and glucose levels. Moreover, in a series of 97 cases of BE caused by different etiologies, the cases that were of an unknown etiology showed better outcomes.3 Similar outcomes were observed in one reported case of a patient who showed no confirmation of an existential etiology.6 However, clinical outcomes in the infectious group are generally worse and they are also associated with higher mortality rates.7
In conclusion, BE is a rapidly progressing disease that shows debilitating consequences. Tissue biopsy should be considered in these cases to help establish and, more importantly, rule out neoplastic and infectious disorders. Our case was treated with all aforementioned treatment options, although there was no confirmation of a specific diagnosis and no initial response to treatment. Following intensive supportive and rehabilitative care, significant improvements were evident.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received August 27, 2017.
- Accepted December 20, 2017.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.