


Abstract
Hypokalemic periodic paralysis (HypoPP) is a relatively rare but treatable disorder caused by mutations in the CACNA1S gene. HypoPP patients may experience paralytic episodes associated with hypokalemia and, infrequently, may develop late-onset proximal myopathy. The paralytic attacks are characterized by reversible flaccid paralysis and, in most cases, spare the respiratory muscles and heart. We report a case of CACNA1S periodic paralysis precipitated by vigorous exercise in a 14-year-old boy who presented with sudden-onset paralysis of both his upper and lower extremities. Laboratory evaluation revealed a markedly low serum potassium level. The patient’s symptoms resolved after correction of the potassium abnormality, and he was discharged with no neurological deficits. Although rare, HypoPP must be differentiated from other causes of weakness and paralysis so that proper treatment can be promptly initiated to ensure good outcomes.
Hypokalemic periodic paralysis (HypoPP) is an inherited disorder caused by autosomal dominant gene mutations characterized by recurrent episodes of hypokalemia and muscle weakness. Symptom onset occurs during the first or second decades of life, with severity varying substantially in terms of the frequency and duration of paralytic attacks. Some patients may experience a single episode in their lifetime; in most cases, however, patients experience repeated crises, which may occur daily, weekly, monthly, or less often.1 HypoPP can be divided into primary and secondary disorders. Two forms of primary HypoPP have been identified: thyrotoxic HypoPP, which is associated with thyrotoxicosis and is more common in adult males, and familial HypoPP, which is a genetic disorder with a lower penetrance in women.1 Although the mechanism underlying the pathogenesis of familial HypoPP is unclear, the condition is associated with mutations in genes encoding subunits of ion channels primarily expressed in striated muscle. These include the voltage-dependent L-type calcium channel subunit alpha 1S (CACNA1S) and the voltage-gated sodium channel type IV alpha subunit (SCN4A).2 During the last decade, a few isolated cases of familial HypoPP have been reported in Saudi Arabia, including thyrotoxic HypoPP in an Asian man3 and another in a Saudi man.4 We present the case of a Saudi teenage boy with HypoPP due to CACNA1S gene mutation.
Case Report
Patient information
A 14-year-old Saudi boy whose medical history was irrelevant presented to the emergency room with sudden-onset paralysis. The patient had gone to bed at 11 p.m. with no weakness and woke up at 6 a.m. unable to move his lower extremities. This was associated with mild pain but no paresthesia; however, during the day, his upper extremities were involved. The weakness was bilateral and involved both the proximal and distal muscles of both the upper and lower limbs.
The day before he presented to our institution, the patient participated in a football game and returned home exhausted. He had been healthy and denied any recent diarrhea, chest pain, shortness of breath, or weight change and had not consumed a carbohydrate-rich meal prior to the episodes of paralysis. He had no history of recent upper respiratory tract infection, fever, vomiting, blurring of vision, headache, dizziness, seizures, loss of consciousness, or swallowing difficulty. He could move his neck and had good facial muscle expressions and eye movements. No episodes of palpitations or chest pain or sweating were reported. He was not on regular medications and did not take any medications before the episode. He denied any history of substance abuse. No new changes in his diet or activity were reported.
He reported experiencing 3 similar episodes, with the first occurring approximately 2 years ago. The patient was examined in a local health center, but no laboratory investigations were conducted as he recovered after intravenous fluid administration. The last episode occurred approximately 6 months ago, and he was examined at the emergency department of our institution. Laboratory investigations showed severe hypokalemia. All 3 episodes were precipitated by vigorous exercise. Eleven of his 12 siblings were all well except for the eldest (36 years old), who had insulin-dependent diabetes mellitus. His parents were consanguineous, but their medical histories were unremarkable. No family history of thyroid disease or a similar condition was reported.
Clinical findings
On physical examination, the patient had a heart rate of 88 beats/minute and a blood pressure of 107/57 mmHg. He weighed 57 kg (75th percentile) and his height was 160 cm (75th percentile). There was flaccid paralysis with power grade 0 in all extremities, which involved the proximal and distal muscles and included the hips and shoulders. Sensation was intact, but deep tendon reflexes were slightly diminished. The rest of the physical examination was normal.
Diagnostic assessment
Laboratory investigations revealed the following: potassium level, 1.3 mmol/L (reference range: 3.5–5.5 mml/L); sodium, blood urea nitrogen, creatinine, and creatine phosphokinase were all within normal laboratory ranges. Complete blood count, coagulation profile, and liver enzyme tests showed normal findings.
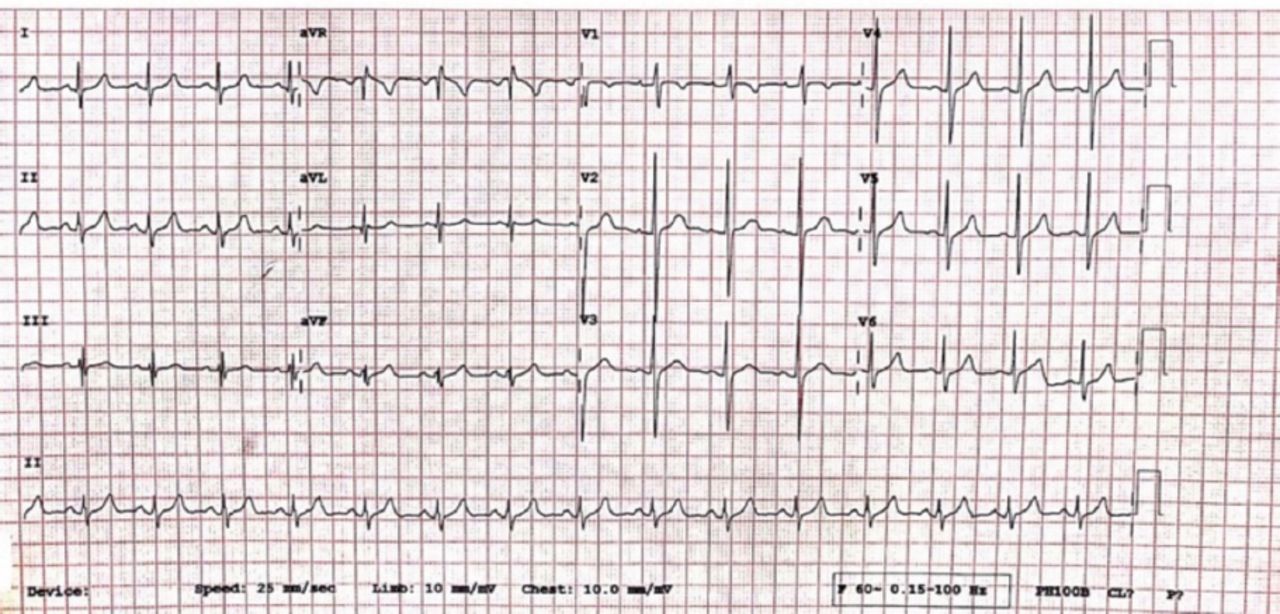
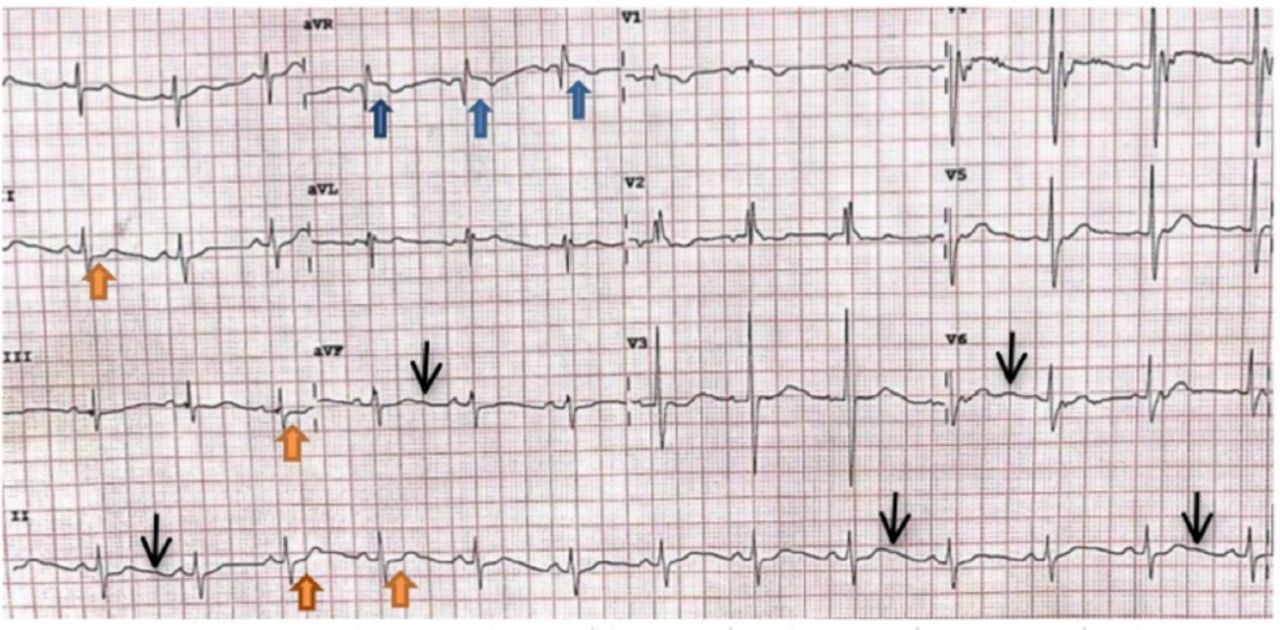
An electrocardiogram showed widespread ST depressions, prominent “U” waves, and ST elevation (lead aVF) consistent with hypokalemia (Figure 1). Computed tomography of the brain showed normal findings.

Electrocardiogram showing ST depression (orange arrows), prominent U waves (black arrows), and ST elevation (blue arrows).
Therapeutic intervention
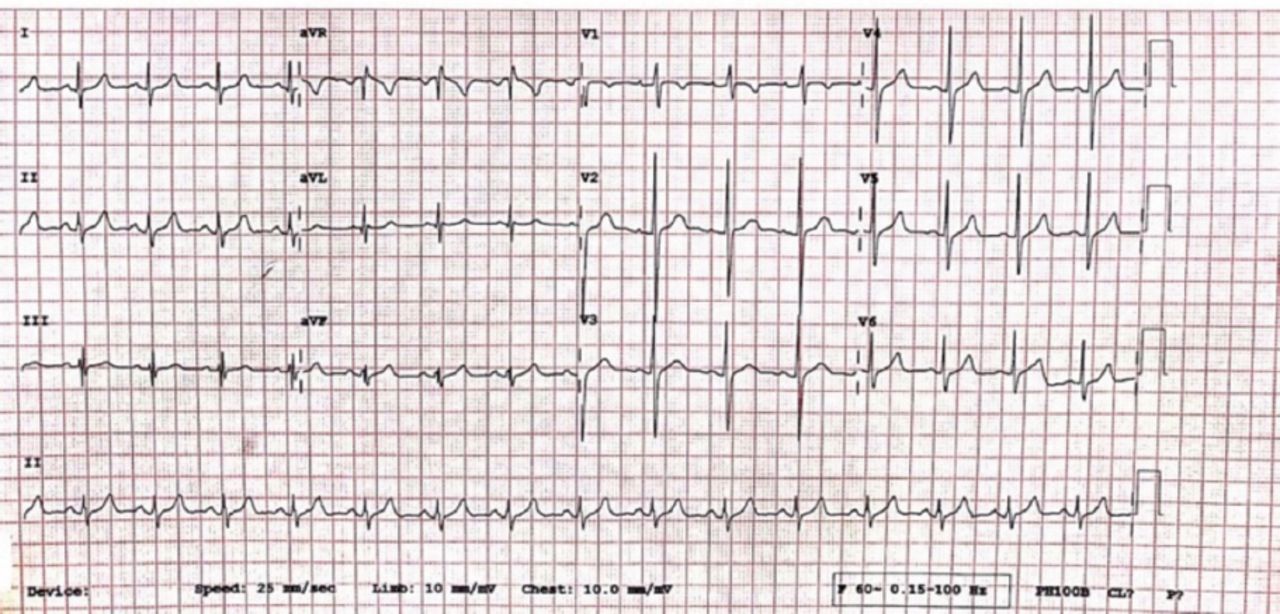
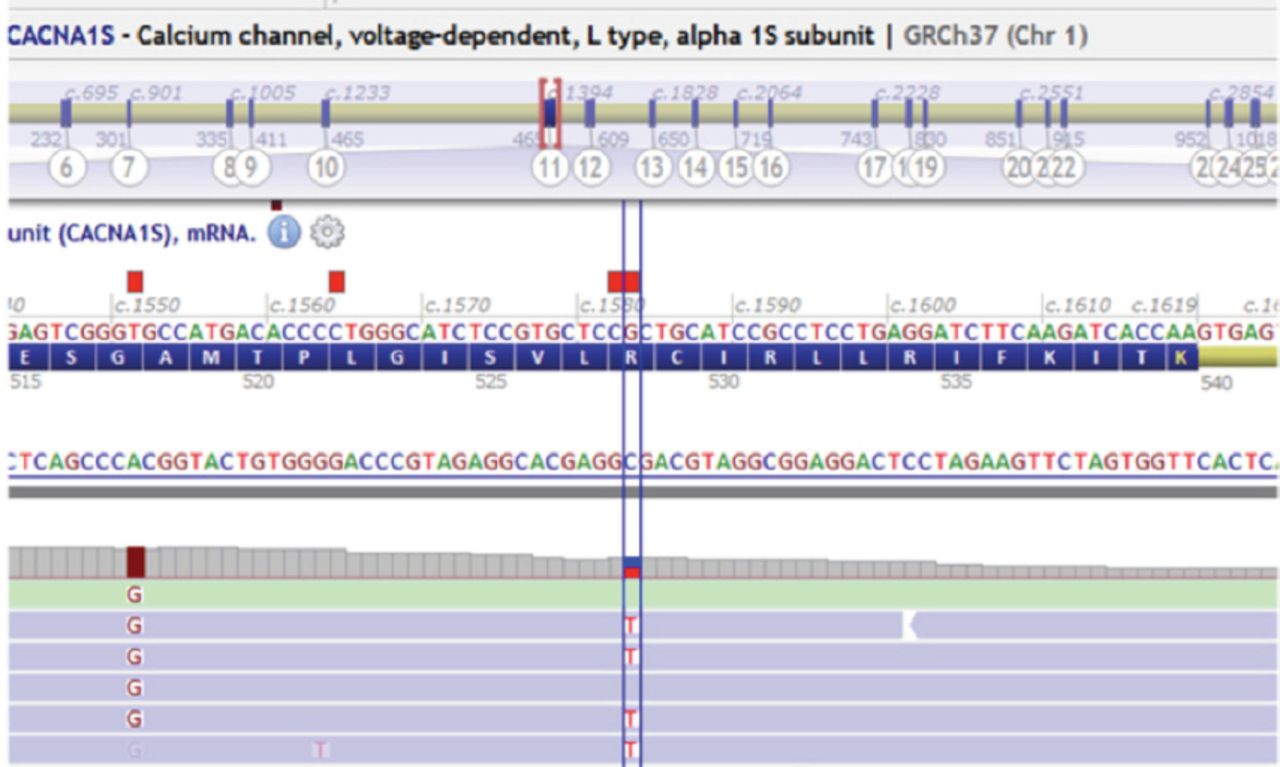
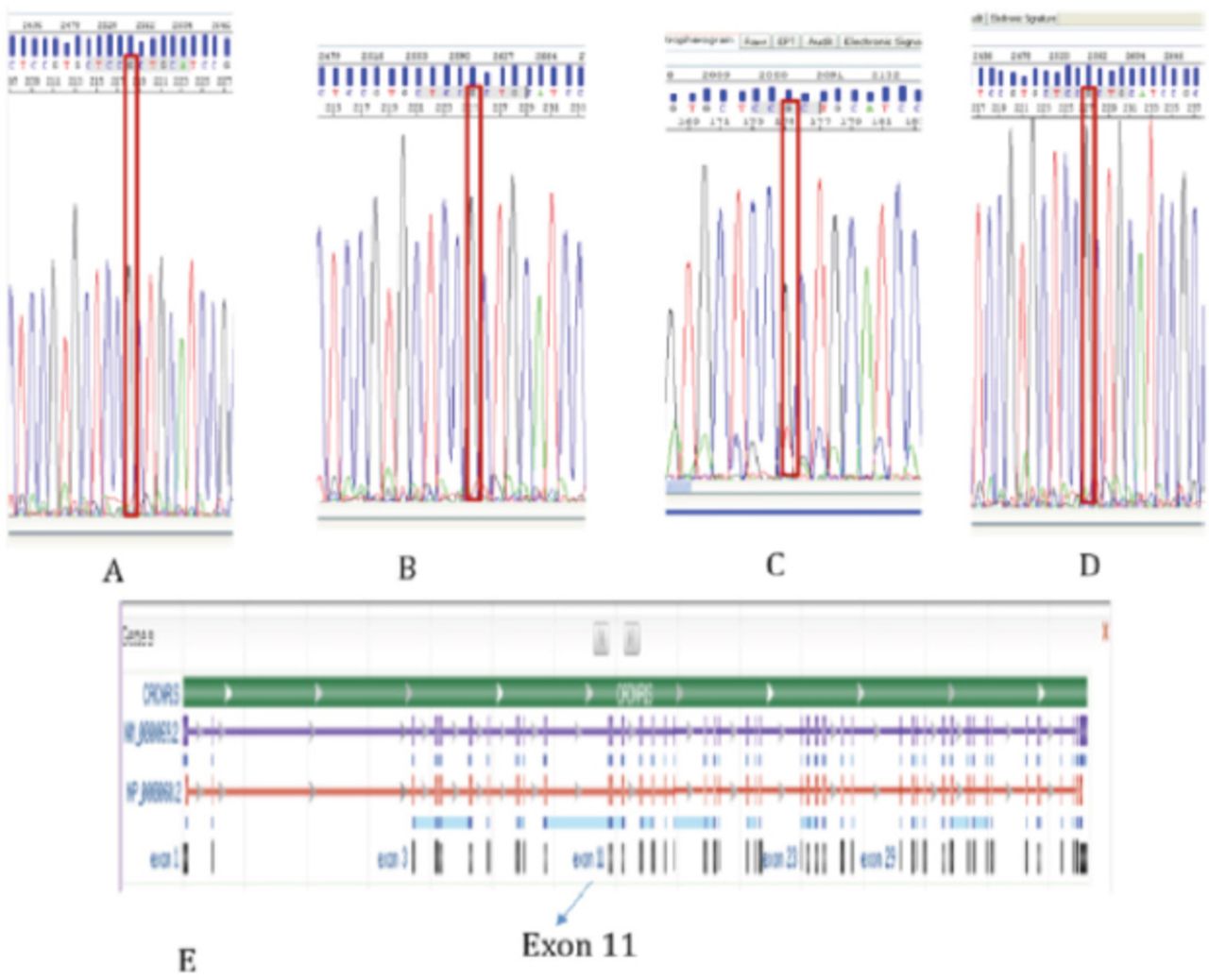
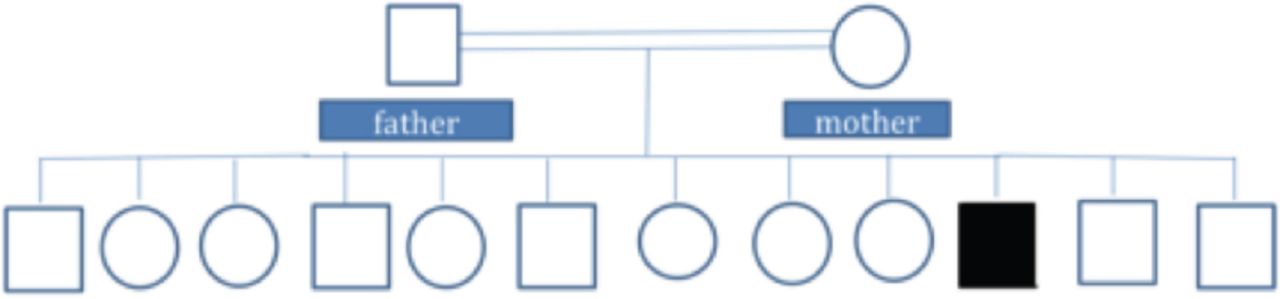
An intravenous potassium replacement was given, then a few hours later, patient’s neurologic symptoms had completely resolved once serum potassium level normalized. A repeat echocardiogram showed normal results (Figure 2). Follow-up studies were performed to determine the etiology of hypokalemia. Urine electrolytes, serum electrolytes, serum aldosterone, and renin levels were measured to rule out adrenal involvement; the results were normal. Thyroid panel results were also normal. Whole exome sequencing revealed a heterozygous pathologic variant in the CACNA1S gene (Figure 3). This was a missense mutation in exon 11 of the CACNA1S gene, c.1583G>A p.(Arg528His), causing an amino acid change from Arg to His at position 528. The genetic diagnosis of autosomal dominant CACNA1S-related disorder was confirmed. Maternal genetic testing was performed using Sanger sequencing, and the results are as shown in Figure 4A. Sanger sequencing testing was also performed in 3 of the patient’s siblings: his sister (Figure 4B) and 2 brothers (Figure 4 C and D). The mother and the tested siblings did not carry the mutation. Testing was not performed in other family members because the patient’s father refused to do the genetic testing for him as well for other older sons and daughters as they were completely asymptomatic. The family pedigree is shown in Figure 5.
Normal ECG after treatment and normalization of the patient’s serum potassium level. ECG - electrocardiogram

Whole exome sequencing result showing the CACNA1S variant, c.1583G>A p.(Arg528His).

Mutation analysis for the sister (A) and mother (B). Mutation analysis for the patient’s brothers (C and D). NM_000069.2 6168 bp DNA refseqgene on chromosome 1 (E). Mutation site at C.1583 G>A (Arginine 528 Histidine) Mutation site at exon 11.
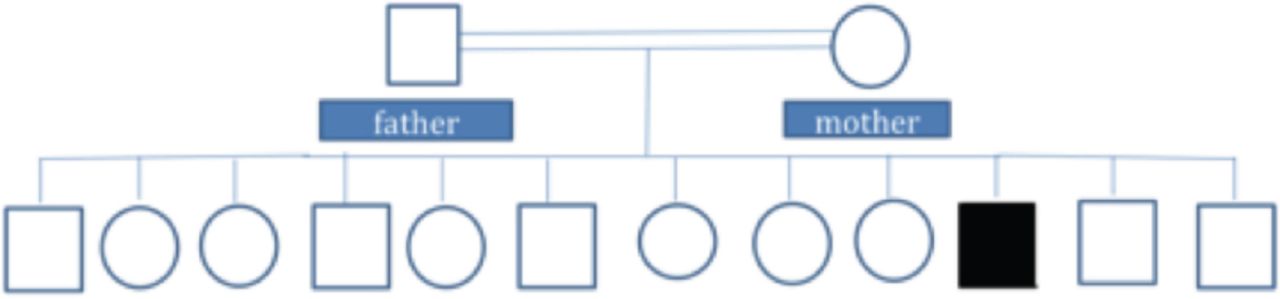
Family pedigree chart. The black symbol represents the patient as diagnosed by genetic tests. Unaffected family members as determined by genetic testing (mother and three siblings) and phenotypic history (father and other older sons and daughters) are indicated by white symbols.
Follow-up and outcomes
The patient was readmitted 2 months after his discharge from the emergency room with the same signs and symptoms. The patient was treated with parental fluid therapy and electrolytes without dextrose, which resulted in the improvement of his symptoms. Oral acetazolamide was administered, and he was discharged. He was examined during 2 subsequent follow-up visits and did not report new episodes of paralysis.
Discussion
HypoPP can severely impact patients’ quality of life. The condition can be more serious in patients with respiratory muscle involvement during paralytic attacks or cardiac dysrhythmia due to hypokalemia.2 The diagnosis of HypoPP is based on the clinical picture and an analogous fluctuation in serum potassium concentrations, lack of other potential causes, and response to treatment,5 as demonstrated in our case. Molecular genetic testing is recommended when the diagnosis of hypoPP is suspected. Sequence analysis has been shown to detect approximately 100% of the pathogenic variants in cases with CACNA1S or SCN4A mutations.6 It is estimated that about 40–60% of HypoPP cases are attributed to CACNA1S mutations versus 7-10% for SCN4A mutations.6
The patient in our report had clinical features of HypoPP that include paralytic episodes associated with concomitant hypokalemia. Most patients typically present with symmetrical lower limb weakness precipitated by the consumption of a carbohydrate diet, recovery from exercise, upper respiratory tract infection, or an emotionally stressful situation.7 Our patient had bilateral weakness of the proximal and distal muscles of both the upper and lower limbs and reported exercising vigorously 24 hours prior to his visit to the emergency room. Although symptom onset is typical in adolescents or young adults with HypoPP, physicians can often miss the diagnosis due to the rarity of the disorder. Physicians might dismiss partial paralysis as secondary to anxiety, especially in younger patients who are otherwise healthy and fit.
Relevant past medical history and interventions of a 14-year-old Saudi boy whose medical history was irrelevant.
In patients with HypoPP, the generally accepted mechanism is that the mutations mostly affect arginine residues in S4 transmembrane helices of CaV1.1 and NaV1.4 channels and cause ions to leak through these transmembrane domains.8 This leads to membrane depolarization, inactivation of sodium channels, and flaccid paralysis. In this patient, genetic testing confirmed an autosomal dominant CACNA1S-related disorder, and he was referred for genetic counseling. Considering the autosomal dominant mode of inheritance, with 100% penetrance in males,9 it is recommended to offer genetic counselling to all patients with familial HypoPP planning to have children. Based on the results of genetic testing, we believe the variant is de novo in this family, as the mother and the tested siblings did not carry the mutation and his father and the rest of his siblings were not clinically affected.
Genetic studies have shown the high rate of occurrence of CACNA1S mutations in patients with HypoPP.10,11 In one report,11 causative mutations were detected in 40 of 58 families with HypoPP. Of these, 40 cases had CACNA1S mutations (69.0%) compared to 5 cases with SCN4A mutations (8.6%). The most common mutations in CACNA1S were described at amino acid positions R528 and R1239, which represented 45.0 and 24.0% of mutations identified, respectively.12 Another study of 83 patients with HypoPP12 revealed a high mutation rate (87.9%), of which 65 (78.3%) were due to CACNA1S mutations.
One of the mainstays of the long-term management is to prevent primary manifestations of HypoPP. The frequency and intensity of paralytic attacks can be reduced by avoiding triggering factors such as vigorous exercise, stress, or carbohydrate-rich meals; adhering to a diet low in sodium and carbohydrate and rich in potassium; and supplementing with oral potassium.2 However, it is often impractical to avoid triggering factors owing to the patient’s social and personal life, as in our patient’s case. Recurrent attacks may cause substantial morbidity and consequently impact the patient’s social, mental, and general well-being.13 Medical treatment with acetazolamide may be necessary to prevent attacks in cases where dietary intervention and oral potassium supplementation do not prove effective.2 Other treatments, such as potassium-sparing diuretics, should be considered in cases where acetazolamide exacerbates attacks. Patients with ongoing paralysis episodes have been shown to benefit from immediate slow infusion of potassium chloride.3,4
The efficacy of medical treatment may differ depending on the specific mutation, albeit further studies should be conducted to investigate these differences. A retrospective analysis of 74 genotyped patients showed that those with CACNA1S mutations were more likely to report an improvement in their symptoms following the administration of acetazolamide than those with SCN4A mutations.6 Approximately 56% of the patients with CACNA1S mutations benefitted from the administration of acetazolamide versus 16% of cases with SCN4A mutations.6
In conclusion, HypoPP should be considered as a differential diagnosis in a patient with sudden-onset paralysis in the absence of risk factors for stroke. It is not uncommon for clinicians to miss the diagnosis due to the variability in the clinical presentation of this condition. Rapid correction of potassium abnormalities can result in the fast and complete resolution of symptoms. When possible, the underlying cause must be adequately addressed to prevent the persistence or recurrence of paralysis, and genetic testing should be performed to confirm the diagnosis.
Acknowledgment
The authors would like to thank Dr. Malak Alghamdi, Department of Pediatrics, College of Medicine, King Saud University, for her critical review.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received September 20, 2018.
- Accepted March 24, 2019.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.