


Abstract
Neuronal ceroid lipofuscinoses (NCLs) are the most common group of neurodegenerative diseases that presents in childhood and are characterized by seizures and progressive neurological deterioration, which results in dementia, ataxia, visual failure, and various forms of abnormal movement. The most common form of neuronal ceroid lipofuscinoses is late infantile (LI-NCL), in association with the genes CLN2, CLN5, CLN6, and CLN8. We report the cases of neuronal ceroid lipofuscinoses type 8 in 3 patients from 2 unrelated families, which was confirmed by molecular testing in 2 of them. Multiple spontaneous abortions, early death, and early onset of motor disability were observed in our cases, reflecting a possible association of NCL 8 with other unrecognized neurodegenerative diseases. Our results expand the genotypic/phenotypic background of variant late Infantile-NCL in Arabic ethnicity.
Neuronal ceroid lipofuscinoses (NCLs) are a group of genetically inherited lysosomal neurodegenerative diseases that mainly presents in childhood.1 The NCL is characterized by permanent cell damage and ultimately multi-organ dysfunction due to intracellular accumulation of abnormal autofluorescent lipopigment materials in multiple cells, including neurons, skin, and skeletal muscle.2–3 Typically, NCL is clinically characterized by both motor and mental regression, seizures, visual impairment, and early death. Historically, NCL phenotypes were classified into 6 categories, based on both the clinical presentation of the patient and their age at onset (congenital, infantile, late infantile, variant late infantile, juvenile, and adult). To date, fourteen NCL genetic forms (CLN1 to CLN14) have been identified, which has led to a new classification system based on the associated gene.2,4 The NCL accounts for 5% of all neurodegenerative disorders in Saudi Arabia.5 The worldwide prevalence of NCL varies among different geographic and ethnic regions, but ranges from 1: 100,000 to 1: 1,000,000 live births.2 The most frequent form of NCL is late infantile NCL (LI-NCL), with a reported incidence of 0.78: 100,000 live births.6 The NCL type 8 can cause two main phenotypes: epilepsy progressive with mental retardation (EPMR), and variant LI-NCL (vLI-NCL). A new congenital CLN8 phenotype characterized by psychomotor retardation and epilepsy from birth was recently described by Pesola et al7 from Argentina. Here, we report the cases of 3 patients from 2 unrelated families who demonstrating the LI- NCL phenotype, 2 of whom were diagnosed through molecular testing to have a homozygous mutation of the CLN8 gene.
Case Report
Patient A. Patient information
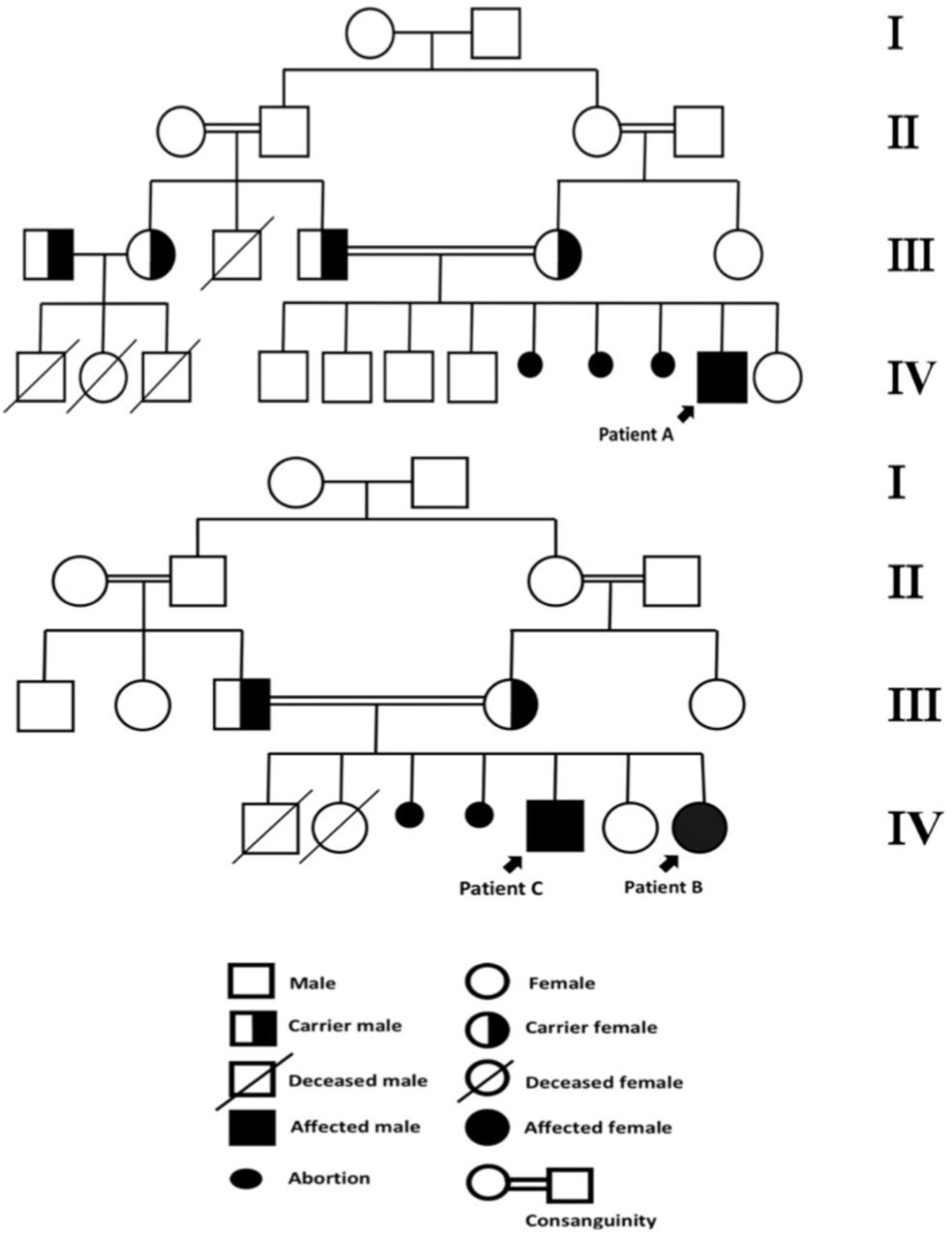
This was the case of a 4.6-year-old Saudi boy, who was the 5th child from a 1st degree consanguineous marriage. He was born full term from an uneventful pregnancy. The child was developing completely normal, until the age of 2 years. Thereafter, he started to display myoclonic seizures that progressively increased in frequency during the subsequent 2 years. By the age of 3 years, he started regressing in cognition, speech, motor, and vision, respectively. Within the last year, he progressively became ataxic, incontinent, and developed spastic quadriplegia. The patient had 5 normal siblings; however, the mother revealed a history of having 3 spontaneous abortions. Furthermore, the patients’ family demonstrated a history of similar phenotypes of neuroregression in four relatives, with the onset of disease ranging between 2 and 4 years of age, and lifespan ranging between 8 and10 years (Figure1). Unfortunately, documentation for these patients was not available.

-Family pedigrees of patients A, B, and C.
Clinical findings
Physical examination of the patient revealed spastic quadriplegia with ataxic movement. The patient had no dysmorphic features or neurocutaneous stigmata. Growth parameters were within normal ranges: height=100 cm (at 10th percentile), weight= 15 kg (at 25th percentile), and head circumference=49 cm (at 10th percentile). Ophthalmologic examination of both eyes revealed pale optic discs.
Diagnostic evaluation
By the age of 4 years, an electroencephalogram (EEG) showed a generalized slow background with polyspike epileptiform discharges; visual evoked potential (VEP) was reduced, and electroretinography (ERG) was normal. Brain magnetic resonance imaging (MRI) revealed signs of cerebellar atrophy. LI-NCL was suspected and confirmed by molecular testing, which showed a homozygous deletion in CLN8 c.(?_-1)_(543+1_544-1)del, which spans the first exon.
Therapeutic intervention
Patient was initially started with valproic acid, and levetiracetam was subsequently added. Daily myoclonic seizures persisted despite maximizing both doses.
Follow up and outcome
Patient was assigned a multidisciplinary team for follow-up care. He was a spastic quadriplegic, and his condition continued to deteriorate rapidly; furthermore, he was not responding to neither the antiepileptic drugs nor physiotherapy.
Patient B. Patient information
Patient B was a 7-year-old Saudi female, and was the 5th child from a 1st degree consanguineous marriage. She was born at full-term from an uneventful pregnancy. The patient demonstrated global developmental delays, particularly with speech and gross motor skills. She was able to sit without support by the age of 18 months, crawl by the age of 20 months, and walk by the age of 24 months. Additionally, she was able to say “mama” and “baba” by the age of 20 months of age, but was unable to speak any further words thereafter. By the age of 2 years, she developed an ataxic gait with frequent falls and myoclonic seizures. Subsequently, she started demonstrating regression in motor, cognition, speech, and vision, respectively. She developed spastic quadriplegia by the age of 4 years. During the subsequent 2 years, she developed generalized tonic-clonic (GTC) seizures, which then became refractory, despite receiving high doses of three antiepileptic drugs. The patient’s family history was significant (Figure1), including having a brother (Patient C) with a similar phenotype. She also had two siblings who died early from unknown causes by the ages of 2 and 4 months. The patient’s mother also revealed a history of having 2 spontaneous abortions with no cause identified.
Clinical findings
Patient B was spastic quadriplegic. She had no dysmorphic features or neurocutaneous stigmata. Growth parameters were within low normal ranges: height=115 cm (at 10th percentile), weight=17 kg (at 3rd percentile), and head circumference= 50 cm (at 25th percentile). Basic funduscopic examination was performed by an ophthalmologist, which revealed severely pale optic discs in both eyes.
Diagnostic evaluation
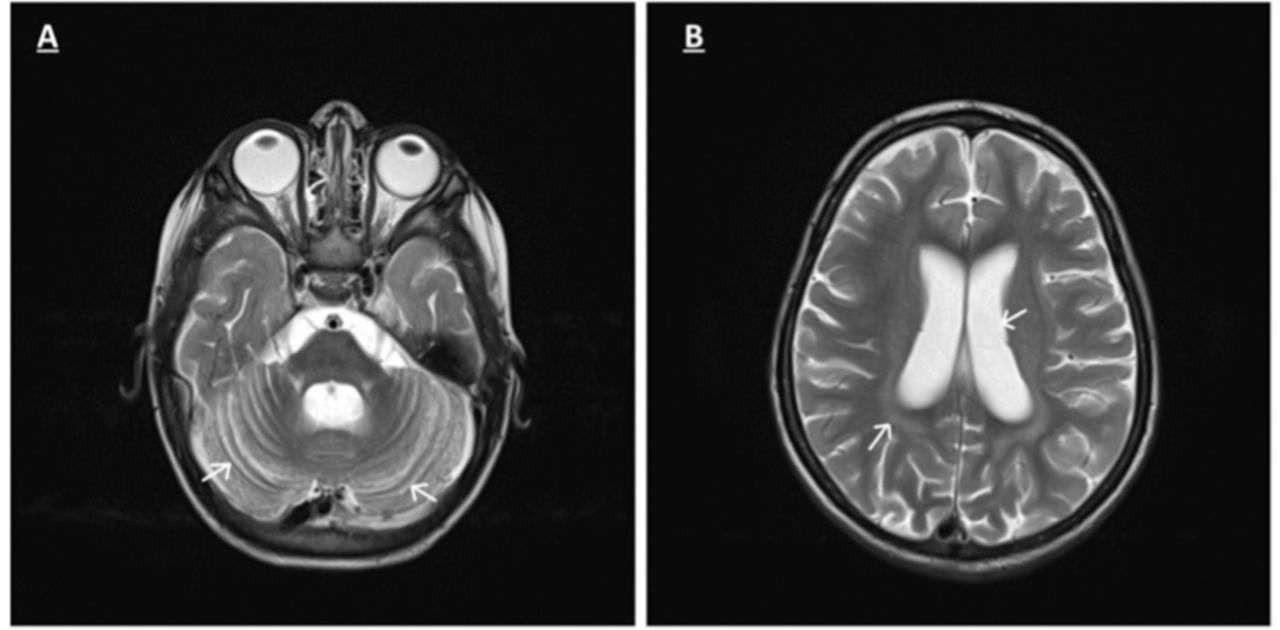
At 3 years-of-age, an EEG showed diffuse slow background activity with generalized epileptiform discharges; ERG was absent, and VEP was reduced. Brain MRI revealed early cerebral and cerebellar atrophy (Figure2). NCL was suspected, and was subsequently confirmed by molecular testing, which showed a homozygous frameshift variant in the CLN8 gene, c.699_700delGT p.Phe234Profs*12, which spans exon 2.

-Axial T2 weighted MRI images A and B) of patient B show enlarging of cerebellum folia, cortical sulci and ventricles with slight hyperintensity of cerebral white matter related to early cerebellar atrophy.
Therapeutic intervention
The patient’s seizures were refractory, despite receiving high doses of three antiepileptic drugs including phenobarbitone, valproic acid, and levetiracetam.
Follow up and outcome
Follow-up-care was provided by a multidisciplinary team; however, her condition continued to deteriorate overtime, and she did not respond to antiepileptics. Patient was frequently admitted for recurrent respiratory infections associated with vomiting and weight loss. Ultimately, gastrostomy tube (GT) feeding was started.
Patient C. Patient information
Patient C was an older brother of patient B (Figure1). He was the 3rd child from this marriage and was born at full-term from an uneventful pregnancy. He was brought to the hospital by his parents following the diagnosis of his sister’s condition, patient B. The child was initially free of any symptoms, demonstrating a phenotype similar to that of patient B. By the age of 7 years, he displayed developmental regression, ataxia with frequent falls, along with myoclonic and GTC seizures.
Clinical findings
The patient was bedridden, severely spastic quadriplegic, and on GT feeding. Growth parameters were within normal ranges. The patient had no neurocutaneous stigmata.
Diagnostic evaluation
By age of 7 years, an EEG was conducted, which showed diffuse slow background activity with generalized epileptiform discharges. Brain MRI revealed cerebellar atrophy. Unfortunately, he died before genetic diagnostic study could be conducted.
Follow up and Outcome
The patient’s condition was progressively deteriorating, and he was not responding to antiepileptics, until he died at the age of 8 years due to recurrent respiratory infections.
Discussion
In this study, we expand the ethnic diversity of NCL type 8 patients, as we report the cases of 3 patients from two non-related families from Saudi Arabia. The patients presented with seizures, ataxia, and neuroregression in speech, cognitive, motor function, and vision. The patients presented here underwent an initial basic screening and a metabolic work-up including complete blood count, electrolyte levels, renal function tests, liver function test, ammonia levels, lactate levels, tandem mass spectroscopy, and urine for organic acid, all of which were negative. Combining clinical and MRI findings, NCL was suspected. Ultimately, molecular testing, for both patients A and B, was performed to confirm the diagnosis. Patient A’s results revealed a homozygous deletion in CLN8. The observed deletion, CLN8 c.(?_-1)_(543+1_544-1)del, spans the first exon, and its estimated genomic breakpoints were chr8:1719131-1719853. On the contrary, patient B’s results revealed a homozygous CLN8 pathogenic gene mutation, C.699-700delGTpPhe234Profs*12 del, which spans exon 2. According to the modified Variant Classification ACMG 2015 guidelines, both variants were classified as likely pathogenic, and a diagnosis was finally confirmed. Combining the clinical presentation and molecular testing, the clinical courses of both patients A and B were similar to vLI-NCL rather than EPMR, due to their age at onset, myoclonic epilepsy, and the rapid regression thereafter. Unfortunately, patient C died before genetic testing could be performed; however, considering the phenotype and disease course of patient B, we speculate that patient C had the same phenotype as his sister, patient B. Our cases showed similar clinical presentations similar to those of other reported vLI-NCL cases, as shown in Table 1, which ranged from cognitive, motor, and speech regression with seizure, ataxia, and visual symptoms, to spastic quadriplegia and the loss of ambulation. Seizures, mainly myoclonic seizures, were the most commonly reported symptom, and visual deterioration was the latest manifestation. Our cases are different from the cases reported by Gao et al. and Beesley et al., in which their patients had dysphagia, while none of our patients did.1,8 Furthermore, our cases, mainly patient B, reported an earlier presentation of clinical symptoms and onset of motor disabilities compared with patients in the other reported cases. Cerebellar atrophy was observed in the brain MRIs of all our patients, which was consistent with the previously reported cases of NCL type 8 (Table1). Here, we highlight the importance of performing an early brain MRI and its findings for the early recognition and diagnosis of vLI-NCL caused by CLN8 mutations. Our study is unique in that, upon reviewing the history of our patients, we observed that multiple spontaneous abortions during the 2nd and 3rd trimesters were reported by both families, and the family of patient B reported early deaths during the first few months of life with no obvious cause, which has not been previously reported to be associated with NCL type 8. With the current study, we raise multiple questions for further investigations and research, to elucidate a possible association between NCL type 8 with other unrecognized neurodegenerative diseases that can explain this phenomenon. In conclusion, these are the cases of vLI-NCL caused by CLN8 reported in Saudi Arabia. Our study expands the genotypic/phenotypic background of vLI- NCL in Arabic ethnicity. We emphasize the importance of early recognition, diagnosis, and counseling in NCL patients, particularly in Arabic regions with a high rate of consanguineous marriages, especially with the recent advances in the use of antisense oligonucleotide medication as a possible treatment modality for NCL type8 in the future.
The clinical and molecular data of our cases and additional selected reported CLN8 cases with LI-NCL
Acknowledgements
We express our appreciation to Dr. Jaafer Al-Obaid, Consultant Neuroradiologist from King Fahad Hospital, Alahsa, Kingdom of Saudi Arabia for his contribution in reading patients’ MRIs. We would like to thank Editage company (www.editage.com) for English language editing.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received October 23, 2019.
- Accepted December 5, 2019.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.