


Abstract
We aim to describe an 8-year-old boy with an unusual clinical presentation of Gaucher disease (GD). Gaucher disease is a progressive lysosomal storage disorder due to deficiency of the specific enzyme glucocerebrosidase with varying clinical features, but often involving the monocytes-macrophages systems. This child ran a progressive course with a devastating outcome. Three distinct GD subtypes have been described with varying clinical features based on the presence or absence of neurologic involvement. Gaucher disease diagnosis is obtained via: enzyme activity assay, gene mutation study, bone marrow aspiration in addition to multiple other tests that have been successfully used in diagnosis of cases of GD. Treatment modalities include enzyme replacement treatment, substrate reduction therapy, bone marrow transplantation, blood transfusion, and surgery are available management modalities for GD. Gaucher disease is a chronic disease requiring a multidisciplinary team approach with regular follow up with multiple subspecialties.
Gaucher disease (GD) was initially described by Philippe Gaucher more than 2 centuries ago, in his doctoral thesis in 1882, when he hypothesized that infiltration of enlarged cells in a spleen represented a “neoplasm.” The biochemical basis for GD was elaborated 83 years later (1965) by Roscoe Brady’s group at the National Institutes of Health. The molecular basis of the disease was elucidated in the late 1980s, when the glucocerebrosidase gene mutations were identified.1 Gaucher disease is characterized by a range of phenotypes, from a perinatal lethal form to an asymptomatic form; however, 3 distinct clinical types have been recognized based on the disease progression and the presence or absence of neurologic involvement.2 Unusual presentation should not be overlooked in this potentially treatable disease, as it may have a positive impact on the disease prognosis and quality of life in the patient.3 Gaucher disease can be diagnosed through enzyme activity assay (EAA), molecular diagnosis, bone marrow aspiration (BMA), and other tests.4,5 Treatment modalities for GD include enzyme replacement treatment (ERT), substrate reduction therapy (SRT), bone marrow transplantation, surgery, and blood transfusion in selected cases.6,7 We aim to describe an 8-year-old boy from consanguineous parents; he had a history of recurrent pyrexia of unknown origin, with equivocal investigations. Raising awareness of early diagnosis of such cases and consideration of unusual presentations of rare diseases can have a positive impact on the progress and long-term management of such cases.
Case Report
An 8-year-old male was referred from a local hospital as a case of meningitis with preceding history of subarachnoid hemorrhage, for which he was previously admitted to the Pediatric Intensive Care Unit (PICU) of the same hospital just prior to his current admission with headache, projectile vomiting, and fever for 2 days. Six months prior to presentation, he had an episode of sudden onset of optic neuritis, which was treated successfully with oral steroids. He had a history of multiple prolonged hospital admissions as a case of pyrexia of unknown origin, without reaching a clear diagnosis. He was previously diagnosed to have alopecia areata at the age of 5 years. Initial assessment upon admission revealed an 8-year-old boy who looked unwell, febrile with a temperature of 39.1°C, pale, not distressed, awake, alert, and conscious without any signs of meningeal irritation. Neurological assessment revealed left-sided hemiparesis. A systemic review revealed distended abdomen with hepatosplenomegaly, and a localized area of alopecia areata approximately 2 × 2 cm in the occipital area. Broad-spectrum parenteral antibiotics and acyclovir were continued according to the plan from the referring hospital. A few hours following admission, he started to complain of severe abdominal pain, persistent fever despite receiving regular analgesics and antipyretics. Initial labs showed normal aspartate aminotransferase (AST), alanine aminotransferase (ALT) with high gamma-glutamyl transferase, high c-reactive protein (CRP), low hemoglobin (HB), low serum iron level with normal unsaturated iron binding capacity (UIBC), and normal HB electrophoresis. In view of the presence of alopecia areata, hepatosplenomegaly, and anemia, the concept of connective tissue disease was raised, which became a remote possibility after the results of antinuclear antibodies (ANA), anti DNA double stranded antibodies, anti cardiolipin immunoglobulin M (IgM), anti cardiolipin (IgG), anti-Sjögren’s-syndrome-related antigen A, anti-Sjögren’s-syndrome-related antigen B came back negative. Stroke work up including, protein C activity, protein S activity, prothrombin time (PT), international normalized ratio (INR), partial prothrombin time (PTT), anti thrombin III (activity), factor V activity were all normal, with absent prothrombin G20210A mutation. Echocardiography was within normal limits. His laboratory workup showed normal lactate dehydrogenase (LDH) with slightly raised ferritin level. Blood film showed moderate anemia, mild microcytosis, and hypochromia with anisocytosis. Normal white blood cells (WBC) count with reactive lymphocytes and monocytes and adequate platelets. Metabolic work up, serum lactate, serum ammonia, plasma amino acids, and urine organic acids were normal. A brain MRI confirmed the presence of right paramedian pontine hyperintense focal area with restricted diffusion, most likely infarction. The pediatric hematologist advised the start of warfarin (Coumadin®; 0.2 mg/kg pars oralis) and a follow up brain CT after 3 days. The pediatric rheumatologist advised to check antineutrophil cytoplasmic antibody (ANCA) levels; proteinase-3 (PR3) cytoplasmic antineutrophil cytoplasmic antibodies, myeloperoxidase perinuclear anti-neutrophil cytoplasmic antibodies were negative. Ophthalmologic consultation revealed normal ophthalmological exam. The dermatologist advised the start of topical treatment for alopecia areata. An abdominal ultrasound revealed mild hepatosplenomegaly with distended gallbladder with innumerable tiny calculi and multiple renal cysts of variable sizes scattering the renal parenchyma bilaterally. The pediatric gastroenterologist advised starting ursodeoxycholic acid. The pediatric nephrologist requested a voiding cystourethrogram (VCUG); which showed no ureteric reflux. A diethylene triamine pentaacetic acid renal scan showed that the right kidney is smaller than left kidney with split renal function: 63% of left kidney, and 37% of right kidney. The impression was that he had adequate function of both kidneys, with no evidence of obstruction, with a large left lower pole cyst. His left sided hemiplegia was improving over time with regular physiotherapy. The erythrocyte sedimentation rate (ESR) and CRP continued rising without the presence of a clear focus, all cultures including blood, urine, and fungal were negative. The pediatric immunologist’s input at that time was this is unlikely to be a case of immunodeficiency. Follow up CT brain after 3 days of commencement of warfarin was free from gross intracranial mass, acute bleed or infarct, no unusual meningeal, or parenchymal enhancement was noted on post contrast study. The pontine hyperintense area that was identified in the previous MRI was not appreciated in the current study by CT criteria.
Early diagnosis
Ten days following admission, he still had a high-grade fever and severe abdominal pain, CT chest with contrast showed few prominent axillary lymph nodes bilaterally. The abdominal CT with contrast showed slightly enlarged liver without focal lesions, and a distended gall bladder with multiple tiny radiopaque densities suggestive of cholelithiasis. The spleen appeared bulky with few sub-capsular wedge shaped areas of low attenuation suggestive of splenic infarcts. Multiple renal cysts were noted bilaterally. The bowel and urinary bladder were unremarkable. There were multiple enlarged mesenteric lymph nodes; more prominent at the mesenteric root region; suggestive of lymphadenitis. This constellation of findings raised the possibility of “GD”. An x-ray of the femur was negative at that time. The pediatric surgeon was consulted for lymph node biopsy and histopathology. Lymph node excision (right groin) revealed reactive lymphoid hyperplasia with features suggestive of dermatopathic lymphadenitis with no evidence of malignancy. He started to suffer from sudden numbness and coldness of his left upper limb, with intact peripheral pulses, ultrasound (US) venous Doppler of the left upper limb showed good flow in the veins with normal responses to proximal and distal augmentation; physiologic reflux and normal cessation of flow was evident with no evidence of thrombosis at this time. A few days later, he started to complain of a painful neck swelling; neck US revealed bilateral benign looking lymph nodes within the jugular chains suggestive of reactive lymph nodes.
The rest of the infectious screen panel revealed negative agglutinin levels to Salmonella Typhi Somatic antigens (O), Salmonella Typhi Flagellar antigens (H), Salmonella Paratyphi types A, B and C Somatic antigens (O), Salmonella Typhi Flagellar antigens (H). In addition, Brucella total antibodies level was negative using Wright agglutination. Helicobacter Immunoglobulin G (IgG) was positive, schistosoma IgG antibody was not detected, toxoplasmsis antibody test was negative. Venereal Disease Research Laboratory was negative. Leishmania antibodies were not detected by immunofluorescence. Purified protein derivative test for tuberculosis was also negative. Epstein–Barr virus (EBV) IgG viral capsid antigen group 125 (VCA gp 125) was positive, VCA p 19 was positive, EBNA-1 was positive, EBV Immunoglobulin M (IgM) VCA gp 125 was negative, VCA p 19 was negative. His clinical picture suggested the late phase of infection. EBV polymerase chain reaction detected a weakly positive EB DNA signal. Cytomegalovirus IgM specific antibody was negative, CMV IgG specific antibodies were positive; denoting previous infection. Malaria smear, human immunodeficiency virus and Mycoplasma pneumonia, IgG, IgM were Negative. Furthermore, no mycobacterial DNA was detected using complex tuberculosis mycobacteria biopsy after gene amplification by PCR. No deleterious mutation detected for familial mediterranean fever.
The CRP continued to increase, with high procalcitonin despite good broad spectrum antibiotic coverage. Normal immunoglobulins level, oxidative burst assay (CGD screening) normal with neutrophil oxidative burst of 1827 (oxidative burst assay by flow cytometry [95% of the patient’s Phorbol Myristate Acetate (PMNs) reduced dihydrorhodamine-123 post PMA stimulations]). Serum copper and ceruloplasmin were normal. High serum ferritin level. Serum uric acid was normal and bone scan was unremarkable.
The patient continued to have continuous severe abdominal pain, with episodes of vomiting. Upper gastrointestinal tract (GIT) endoscopy showed chronic active duodenitis with chronic inactive gastritis, no H. pylori organisms was isolated. His condition deteriorated, and he developed respiratory distress, he was transferred to the PICU, where chest x-ray showed pulmonary edema and congestion, he required intubation, and was connected to a mechanical ventilator, with a nasogastric tube inserted. A few days later, while still in the PICU, he started to have cold extremities, with color changes in his lower limbs with darkening of tips of his toes. A vascular surgeon was consulted, and arterial Doppler US of left lower limb failed to demonstrate blood follow in the posterior tibial and dorsalis pedis arteries. Ultrasound arterial Doppler of both upper limbs showed absent flow in both radial and ulnar arteries bilaterally. His chest condition was stable over time, with persistent pulmonary oedema, follow up abdominal ultrasound showed mild amounts of free fluid at both para-colic gutters and pelvic region, other similar findings to the previous scans. Liver enzymes and serum lactate started to rise. A few days later, interval reduction in bilateral peri-hilar alveolar opacities was noted, though left basal atelectasis was noted in his follow up chest x-ray. His general condition deteriorated with blood culture and sensitivity (C/S) grew multi drug resistant Acinetobacter baumannii, treated with colistin (Colistin ®5 mg/kg/day IV). He started to develop ascites and was found to have hypoalbuminemia; C/S ascitic fluid multi drug resistant type Acinetobacter baumannii was isolated, only sensitive to colistin. A follow up chest x-ray showed interval development of left mild to moderate pneumothorax with partial lung collapse noted and clear right lung. Laparoscopy showed gangrene at various levels, so gut resection, and anastomosis was carried out with ileostomy. Liver biopsy and pathological examination using periodic acid–Schiff stain showed uneven distribution, suggesting a metabolic storage disease. Abdominal ultrasound post laparoscopy showed a large thin walled focal collection in the lesser sac causing mass effect over the stomach with small collections in the right iliac fossa and left lumbar regions noted with trace free fluid in the peritoneal cavity, there were collapsed stomach and bowel loops. Later on, the lesser sac collection was drained through US guidance, and approximately 100 ml of purulent fluid was drained; no residuals were detected by follow up CT scan.
Confirmed diagnosis
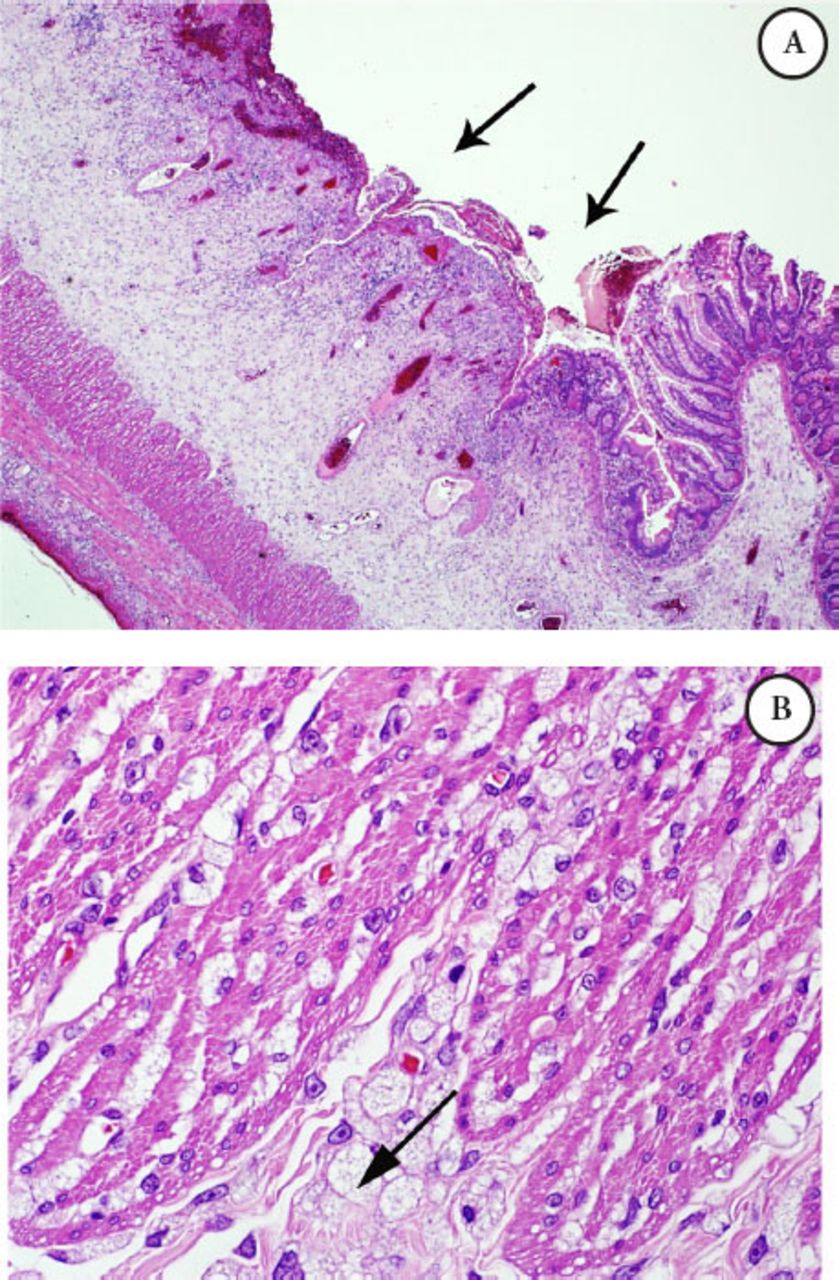
Intestinal biopsy: The bowel has scattered foamy macrophages, which were stained with cluster of differentiation 68 (CD68) namely, CD 68 positive, that form dense clusters mainly around the perforation site, noted in the rest of bowel wall as well, with extensive gangrenous changes in the small bowel, PAS was negative, Alcian blue (mucous stain) was faintly positive, such foamy cells are suggestive of a metabolic disease; biopsy was not suggestive of vasculitis. Microscopic examination of the small bowel showed trans-mural infiltration by sheets of foamy macrophages (Figures 1 A-B & Figure 2).
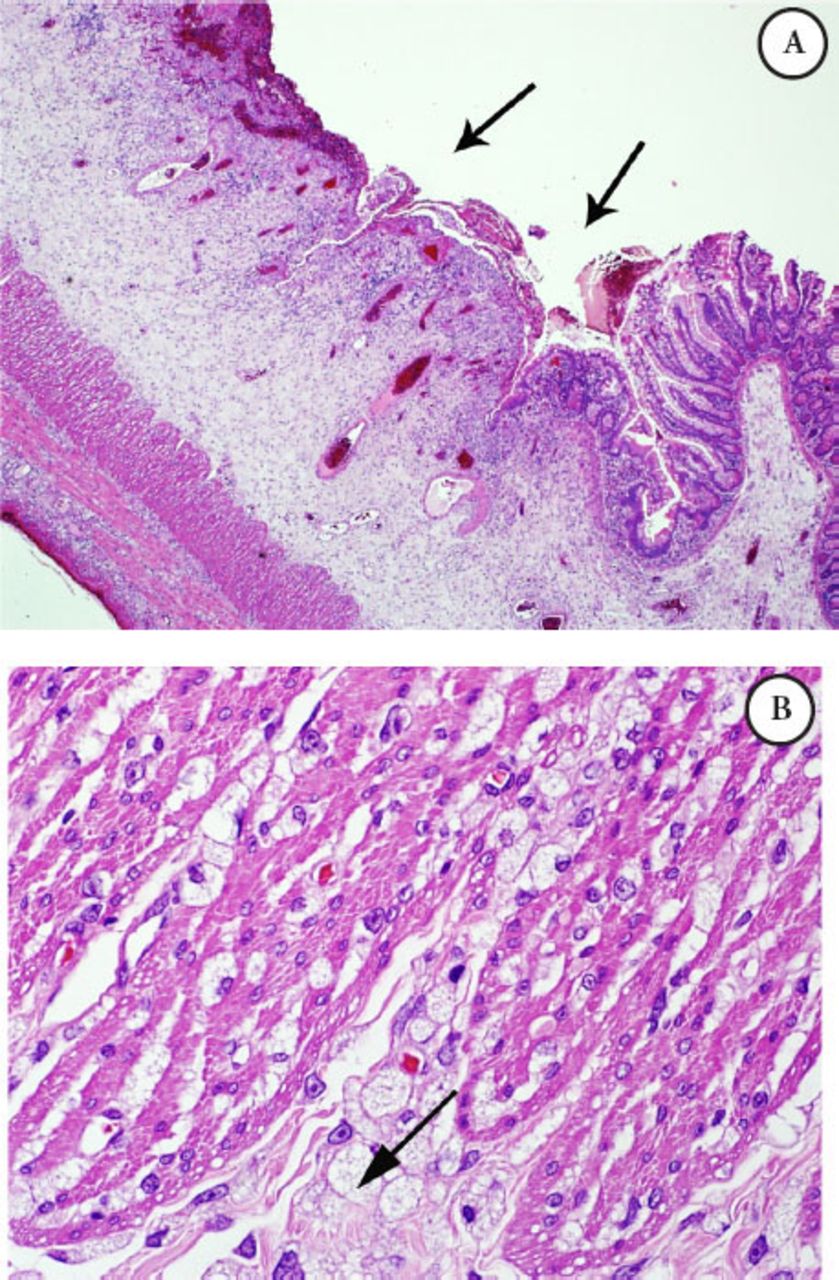
Microscopic examination of the small bowel A) Illustrating mucosal ulceration and expansion of the submucosa, B) Higher power field (40X) exhibiting diffuse infiltration by foamy macrophages with vacuolated cytoplasm intersecting the muscle coat fibers (Gaucher cells).
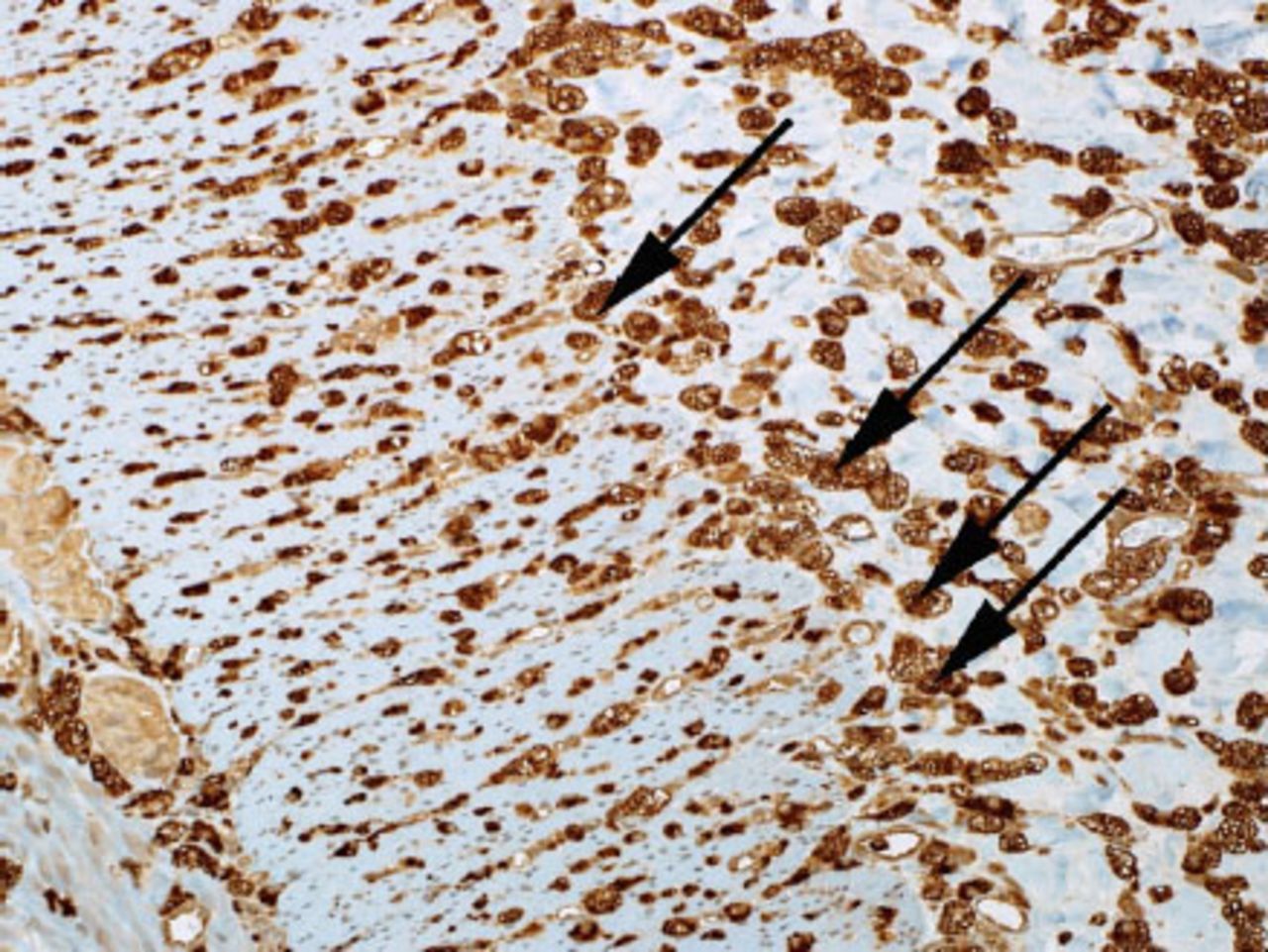
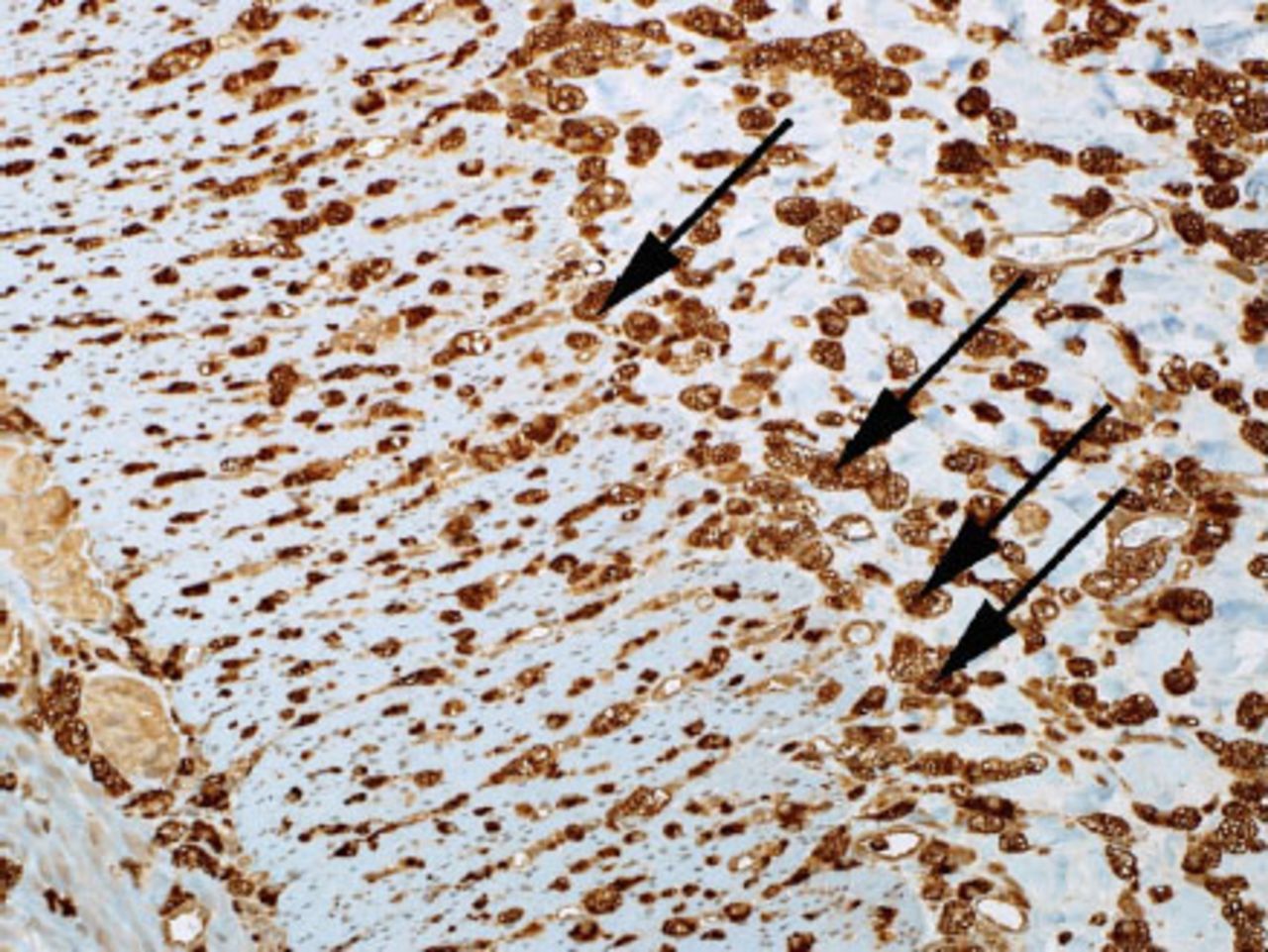
Microscopic examination of the small bowel showing foamy macrophages stained with CD68. (CD-68 is a glycoprotein that binds to low density lipoprotein)
Serum level of Beta Glucocerebrosidase enzyme, 1.3 (4.8-8.9 U) with reference enzyme in the normal range, proved deficiency of B-Glucocerebrosidase, suggesting that the patient is most probably affected by GD. He started to have persistent ascites, extensive gangrene in his fingers and toes, with thrombocytopenia, not responsive to platelet transfusions, Wound culture revealed moderate growth of Acinetobacter baumannii MDR type and Proteus vulgaris, ileostomy, was kept in situ. He started to show a deteriorating chest condition with airspace opacity in the right hilar and para-cardiac region areas this was followed by episodes of disturbed levels of consciousness, urgent CT brain showed multiple different sizes innumerable diffuse foci of hemorrhage scattered through the entire brain (cortical, sub cortical deep parenchymal, and basal ganglia) also with evidence of focal relatively large (3.5×1.7×2.5 cm) blood collections seen intra-parenchymally in the left posterior sub cortical parieto-occipital region with marked surrounding edema with mass effect and effacement of the adjacent sulci (possibly coalesced focal hemorrhage), highly suggestive of the serious condition of hemorrhagic septic emboli. He developed progressive ascites, limb gangrene, and respiratory failure; he passed away a few days later.
Discussion
Gaucher disease is a rare metabolic disorder and a sphingo-lipidosis (a subtype of lysosomal storage disease) that is inherited as an autosomal recessive metabolic disorder that results from deficiency of D-glucosyl-N-acyl sphingosine gluco-hydrolase (also termed glucocerebrosidase, beta-glucosidase). It is chronic and progressive in its clinical presentation, and affects 1 in 60,000-100,000 births worldwide, although pan-ethnic in distribution, it occurs with increased prevalence among the Jewish people of Eastern and Central European descent (Ashkenazi Jews) with an 8.9% carrier rate. It is encoded by the human GBA 1 gene. The GBA 1 gene is located on chromosome 1 q2. There are over 300 known mutations that can cause GD, the most common are c.1226A>G (N370S), 84GG, IVS2+1, and the c.1448T>C (L444P) mutations. The L444P mutation homozygous state has a very high association with neuropathic variants of GD.3
The beta glucosidase is needed to hydrolyze the B-glucosidic linkage of the glucosylceramide, and the deficiency leads to an accumulation of glucosylceramide and other glycolipids within the lysosomes of the mononuclear phagocytes system, especially tissue macrophages, which become engorged and are typically found in the liver, spleen, and bone marrow and occasionally in lungs, kidneys, and intestine. Kupffer cells in liver, osteoclasts in bone, alveolar macrophages in lungs, microglia cells in the CNS can be involved.2
Secondary hematological sequelae include severe anemia and thrombocytopenia in addition to the characteristic progressive hepatosplenomegaly, skeletal complications, including osteonecrosis and osteopenia with secondary pathological fractures. Gaucher cells that are 20-100 mm in diameter are macrophages that are pathologically laden with glycolipid materials particularly in bone marrow; and are the pathohistological hallmark of the disease.3
Gaucher disease is characterized by a range of phenotypes, from a perinatal lethal form to an asymptomatic form; however, 3 distinct clinical types have been recognized based on the disease progression and the presence or absence of neurologic involvement. Each type has been linked to a particular mutation and around 300 mutations are known. Intermediate phenotypes do occur particularly between types 2 and 3, and there have been several reports of several neurological manifestations in type 1, although it is typically classified as non-neuronopathic. The glucocerebrosidase mutations have been associated with a variety of Parkinsonian phenotypes, the mechanism underlying this association remained unclear, though recent evidence suggests a link between GBA 1 gene mutation and the development of Parkinson disease. Definite diagnosis is made by glucocerebroside assay. Molecular studies for mutation are useful for confirming diagnosis, screening family members, and formulating the prognosis.4
Type 1 - non-neuronopathic or the adult form is the most prevalent especially in individuals with Ashkenazi Jewish ancestry. There is a variable age of onset with marked variability among different ethnicities; hence, the frequency is difficult to quantify. Typically, the mean age of presentation is in the late 20s, as an isolated hepatosplenomegaly with mild to moderate effect, although patients may remain asymptomatic throughout life.1
Type 2 - infantile (acute) onset, is an acute neuronopathic form that is rare and the most clinically heterogeneous, rapidly progressive, and more severe with typical age of presentation before 6 months. The brainstem is severely involved with fatal outcome by 2 years of age.5
Type 3 - chronic or subacute (Juvenile or Norrbottnian) neuronopathic form with subtle non-neurological involvement, may present with oculomotor apraxia and seizures. Patients typically survive into the fourth decades of life.5
General clinical features
Gaucher disease should be considered in children of all ages presenting with visceromegaly, recurrent infections, bone pain, fatigue, and thrombocytopenia. Its acute infantile form is characterized by failure to thrive (FTT). Splenomegaly is the most common presenting sign of the non-neuronopathic form, and hepatomegaly is universal but not the same magnitude as splenomegaly. Hepatic fibrosis, cirrhosis, and portal hypertension are uncommon except in patients with prior splenectomy during childhood. Patients with type 2 GD have an earlier onset of bulbar palsy and spastic quadriparesis with regression of developmental milestones. Fatality before 2 years of age is very typical. Type 3 GD is a heterogeneous multisystem and a progressive neuronopathic disorder that although progresses more slowly than type 2 should still be considered in any child presenting with visceromegaly, bone pain fatigue, thrombocytopenia due to splenic sequestration and bone marrow infiltration, and may result in occasional life threatening hemorrhagic diathesis. Anemia is frequent but not severe. Leucopenia is rare, and may be a complication of hypersplenism with easy bruising and epistaxis, short stature, and pubertal delay. Interstitial lung disease and pulmonary hypertension are occasional manifestations. Bone involvement may present with bone pain due to avascular necrosis or pathological fracture due to osteopenia. Another form of GD is the cardiovascular type with aortic and mitral valve calcification and also a perinatal lethal form (very severe) that presents with hydrops fetalis (prenatally), ichthyosis, hepatosplenomegaly, neurological symptoms, and distinctive facies.
Diagnosis
Carrier screening of individuals of Ashkenazi Jewish heritage for prenatal diagnosis through blood or saliva test is reliable one. Diagnostic delays are not unusual despite availability of many non-invasive tests.5 1) Enzyme activity assay (EAA): Measurement of acid-beta glucosidase activity in peripheral blood leukocytes, urine or cultured skin fibroblast. Glucosidase activity of less than 15% is diagnostic. The EAA is sensitive, specific, and much less invasive, it can be carried out prenatally. 2) Molecular diagnosis: DNA analysis of the GBA gene for the 4 most common mutations that account for most of the disease. Gene mutation study for (IN 370 S, L 444 P, 84 GG, IVS2) accounts for approximately 90% of the disease causing alleles. Can be carried out prenatally. 3) Bone marrow aspiration findings of aggregates of large Gaucher cells. Not necessary to establish a diagnosis as Gaucher cells can be sparsely distributed and give false negative bone marrow aspirate. 4) Other tests: Complete blood count (CBC) showing thrombocytopenia, peripheral smear shows microcytosis, hypochromia. Liver function test (LFT), EEG, brainstem-evoked potential, chest x-ray, DEXA scan, ECG, ultrasound of the abdomen, MRI or CT scan of the bone, liver, and spleen, angiotensin converting enzyme (ACE) level and chitotriosidase level.
Treatment. Enzyme replacement treatment (ERT)
The ERT at present is very expensive and lifelong and should be the main core of any therapeutic plan. Due to low profitability and high expense of developing these therapies for this rare condition, the ERT for GD have been granted orphan drug status in the USA in 1994. It has shown to be effective in reversing the hematologic and visceromegaly manifestation. Skeletal symptoms may be slow to respond.7 For type 1 and most type 3 GD, intravenous ERT with recombinant glucocerebrosidase is given every 2 weeks or every other week at a high dose. They are effective in reducing the liver and spleen size, ameliorating skeletal abnormalities and hematological abnormalities of the disorder, although lung manifestation may not respond satisfactorily to ERT. The US FDA approved Imiglucerase alfa (Genzyme Corporation, Oxford, Oxfordshire, UK) in May 1994 for long-term ERT for pediatric and adult patients with a confirmed diagnosis of Type 1 GD,6,7 and Velaglucerase alpha (Shire Human Genetic Therapies, Inc, Basingstoke, Hampshire, England) in February 2010 for the long term treatment of type 1 GD in both the pediatric and adult population and recently Taliglucerase alfa (Pfizer Inc, New York, NY, USA and Protalix Bio Therapeutic Inc. Carmiel, Israel) in May 2012 as a long-term enzyme replacement treatment for type 1 GD.
Substrate reduction therapy
The glycolipid accumulation is prevented by reducing the substrate level. Glucosylceramide synthase, Miglustat (N-butyldeoxynojirimycin-Oxford GlycoSciences Oxford, UK) inhibits glucosylceramide synthase that catalyzes the first step in the biosynthesis of glucosylceramide and subsequently reduces the biosynthesis of more complex glycosphingolipids. It is indicated in patients who are unsuitable for ERT.7
Bone marrow transplantation
May be considered when access to expensive ERT is not possible. May be an effective treatment for neurological manifestation of GD. It can also reverse the non-neurological manifestation of type I, but is rarely performed due to a high risk of significant morbidity and mortality.
Surgery
Splenectomy may be required on rare occasions especially prior to the advent of ERT to ameliorate hypersplenism. Infection with encapsulated bacteria is a complication, and acceleration of other manifestations of the GD like glycolipid deposition in the lung and bone marrow are well-known adverse effects of splenectomy
Blood transfusion
Indicated in some anemic patients. The goal is to increase hemoglobin level to 11 g/dl or more in children with GD. Coexisting etiologies for anemia must always be evaluated and ruled out.
In conclusion, cases of GD can be challenging, with various unclear presentations, initial investigations may not be revealing, and partial response to routine measures may result in significant delay in reaching diagnosis. Many non invasive tests are now available, though blood transfusion prior to enzymatic assay testing can result in false negative results, it is crucial to withdraw the sample prior to transfusion. Prenatal diagnosis is available; proper genetic counseling should be offered to all parents of cases with GD. Early diagnosis and management can result in better quality of life for the affected patient, and can reverse non-neurological complications, though lifelong management is paramount.
Acknowledgments
We gratefully acknowledge the parents of our case, who consented to publication.
- Received October 12, 2014.
- Accepted March 23, 2015.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.