


Abstract
Subependymal giant cell astrocytoma is a benign WHO grade I intraventricular tumor arise in patients with tuberous sclerosis complex. Previous reported described histopathological predictors of more aggressive forms, terms atypical SEGA in infantile age group. Other reports showed possible transformation of SEGA into glioblastoma, or misdiagnosis as glioblastoma due to the presence of atypical histopathological features. Here, we report a case of an infant who presented with right frontal extraventricular SEGA and underwent craniotomy with complete resection. Eight months later, he presented with fast recurrence in same location with midline shift and subfalcine herniation. Histopathological description showed high grade features including Ki labeling index of 60%, atypical mitotic figures, cellular plemorphism and necrosis. We also discussed the possible presence of different entity (termed atypical SEGA) which may have more aggressive clinical course, with literature review of predictors of SEGA aggressiveness and possible transformation/misdiagnosis as glioblastoma.
Tuberous sclerosis complex (TSC) is a multi-systemic neurocutaneous genetic disorder characterized by multiple lesions in skin, brain, eyes, heart, kidneys and lungs. The majority of the tumors in tuberous sclerosis, including facial angiofibroma, subependymal giant cell astrocytoma (SEGA), cardiac rhabdomyoma, renal angiomyolipoma, are benign.1 Malignant tumors in tuberous sclerosis, such as renal cell carcinoma and malignant angiomyolipoma, are extremely rare. In addition, de novo high-grade glioma or malignant transformation of SEGA in tuberous sclerosis is an extremely rare phenomenon.2 Here, we are reporting a case of malignant SEGA in an infant along with a review of the literature.
Case Report
Patient information and clinical examination
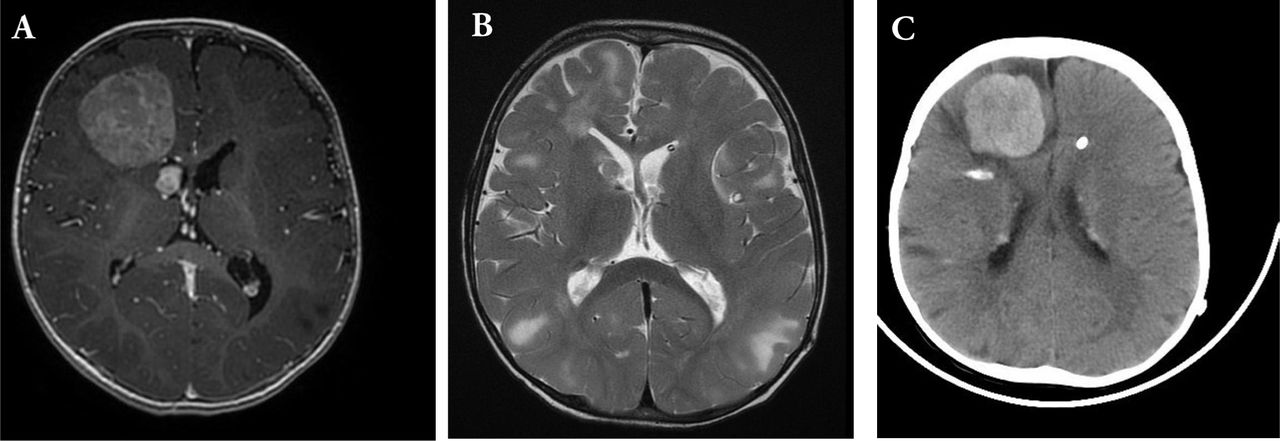
An 11-months-old child diagnosed with TSC presented to our Emergency Department with repeated generalized tonic-clonic seizures (Figure 1). Clinical examination was unremarkable apart from delayed cognitive skills. There was no focal deficits or cranial nerve palsies. Magnetic Resonance Imaging (MRI) of the brain showed multiple cortical tubers that appeared as T2 signal changes with multiple subependymal nodules and a large right frontal enhancing tumor, with areas of calcification (Figure 2A). There was another 1x1 cm periventricular lesion close to the first one.

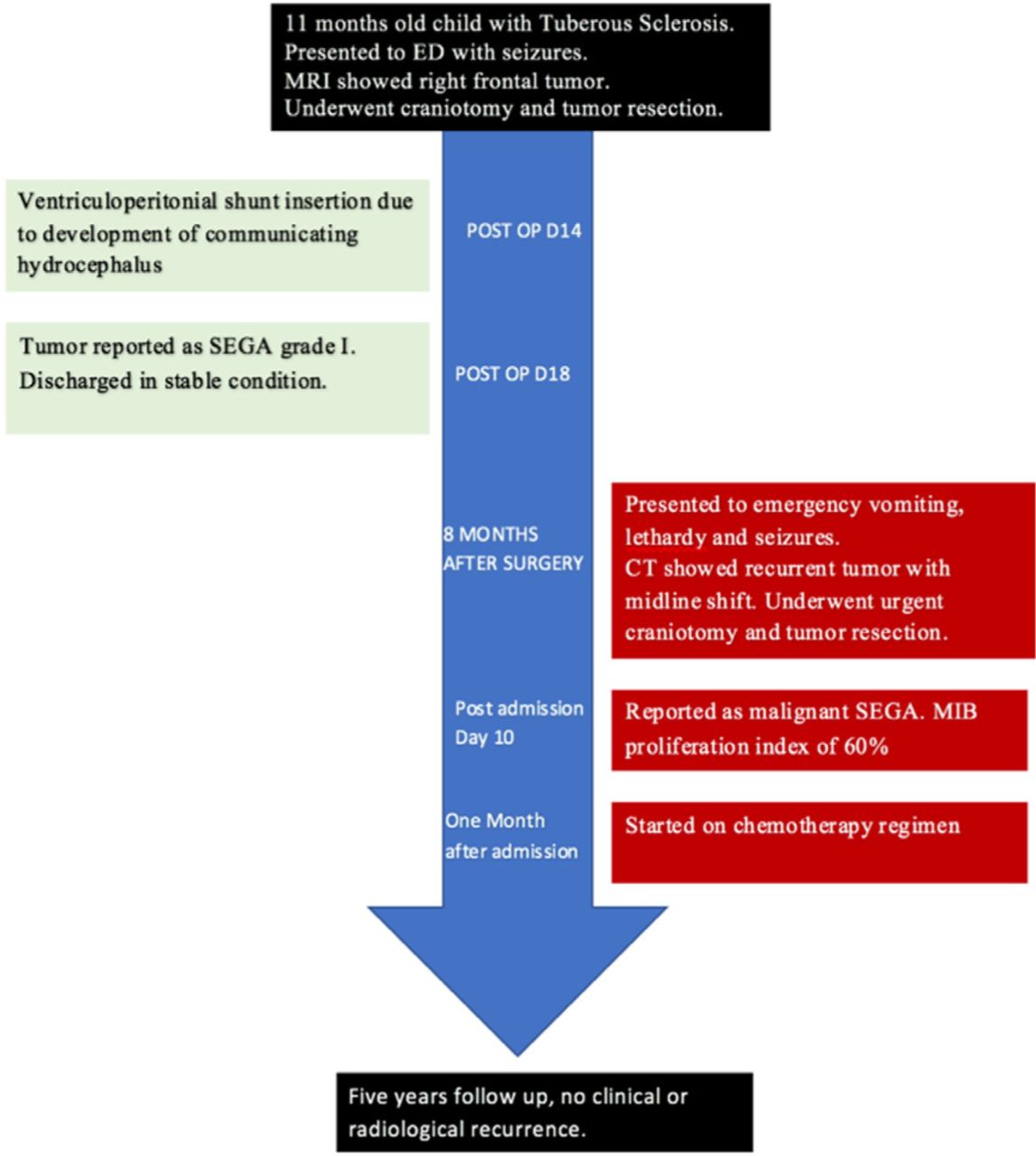
Showing patient hospital course and outcome after discharge.
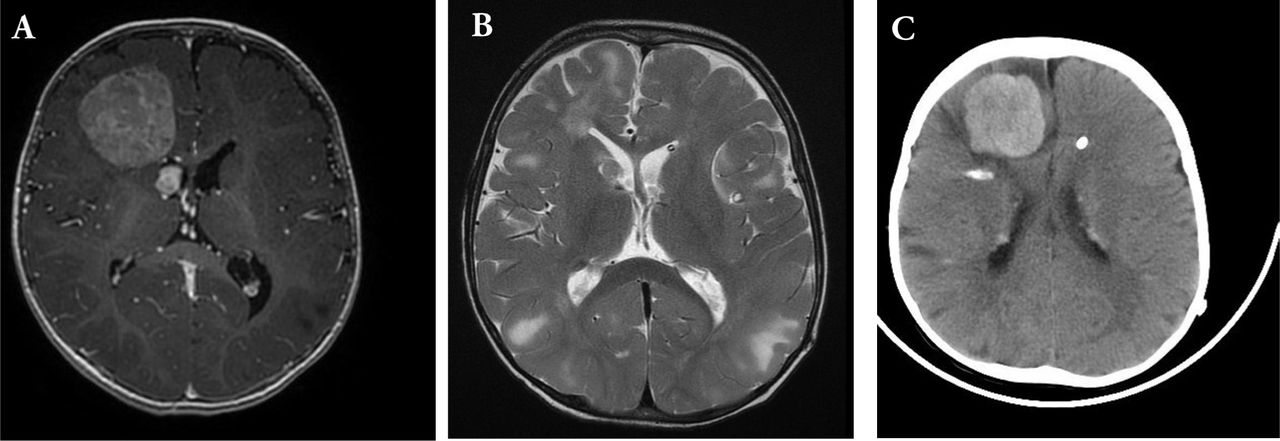
Magnetic resonance imaging T1 with gadolinium, showing A) right frontal homogenously enhancing tumor. Notice the other small right periventricular lesion abutting the right frontal horn. B) Post-operative T2 MRI showing complete resection. C) Recurrence after 8 months. CT brain showed right frontal hyerdense lesion with midline shift and subfalcine herniation. There are areas of calcifications and periventricular hyperdense nodules.
Therapeutic intervention
The patient underwent right frontal craniotomy and gross total resection of the frontal tumor. Histopathological examination showed SEGA WHO grade 1. Post-operative MRI showed complete resection of the tumor (Figure 2B). The patient had ventriculoperitoneal shunt due to postoperative communicating hydrocephalus and was then discharged in stable condition. Eight months after the surgery, he presented to emergency room with irritability, vomiting, lethargy and generalized tonic-clonic seizures. Computed tomography of the brain showed right hyperdense frontal tumor with perilesional edema, hemorrhage, midline shift and subfalcine herniation (Figure 2C). He underwent emergency right frontal craniotomy and gross total resection of the tumor.
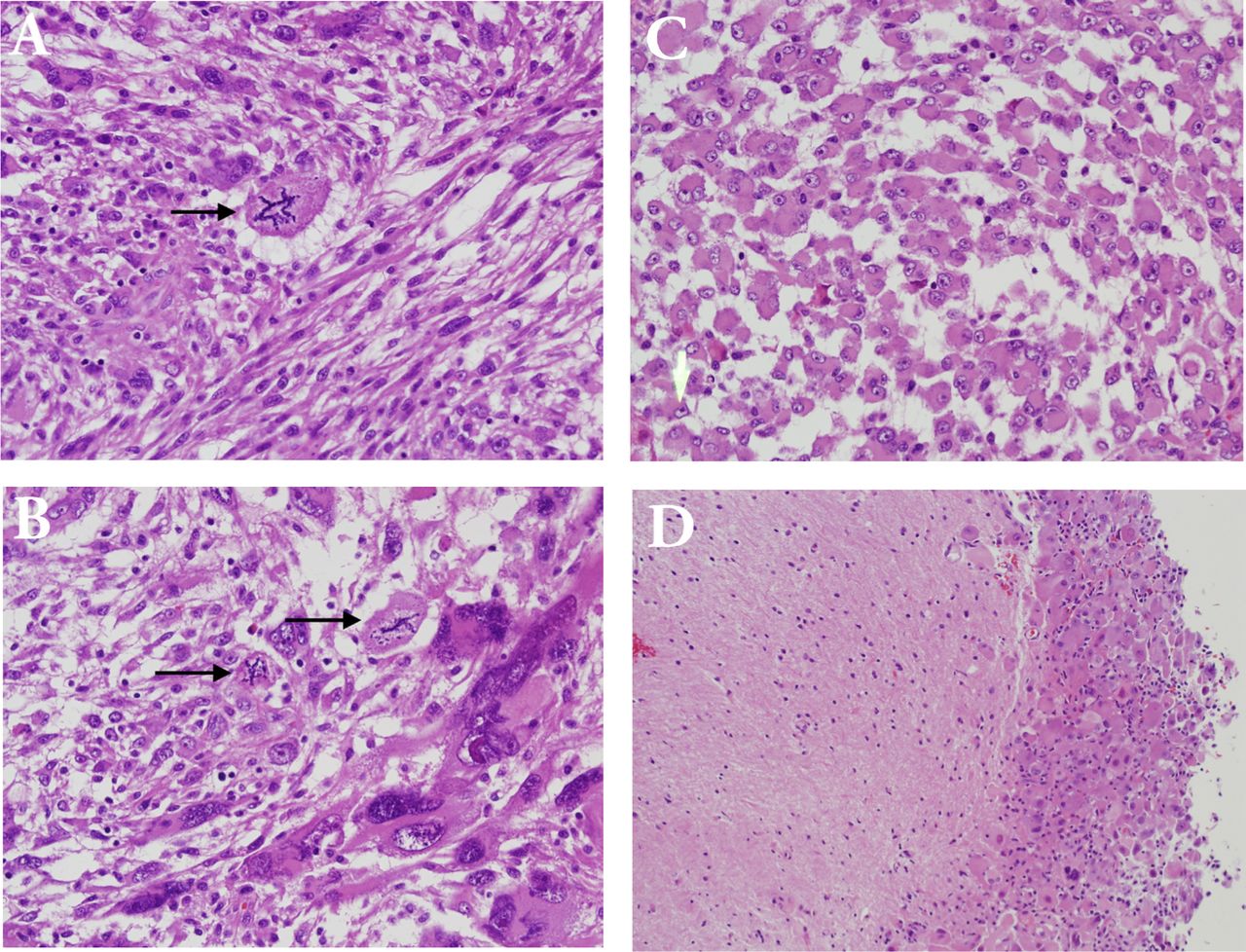
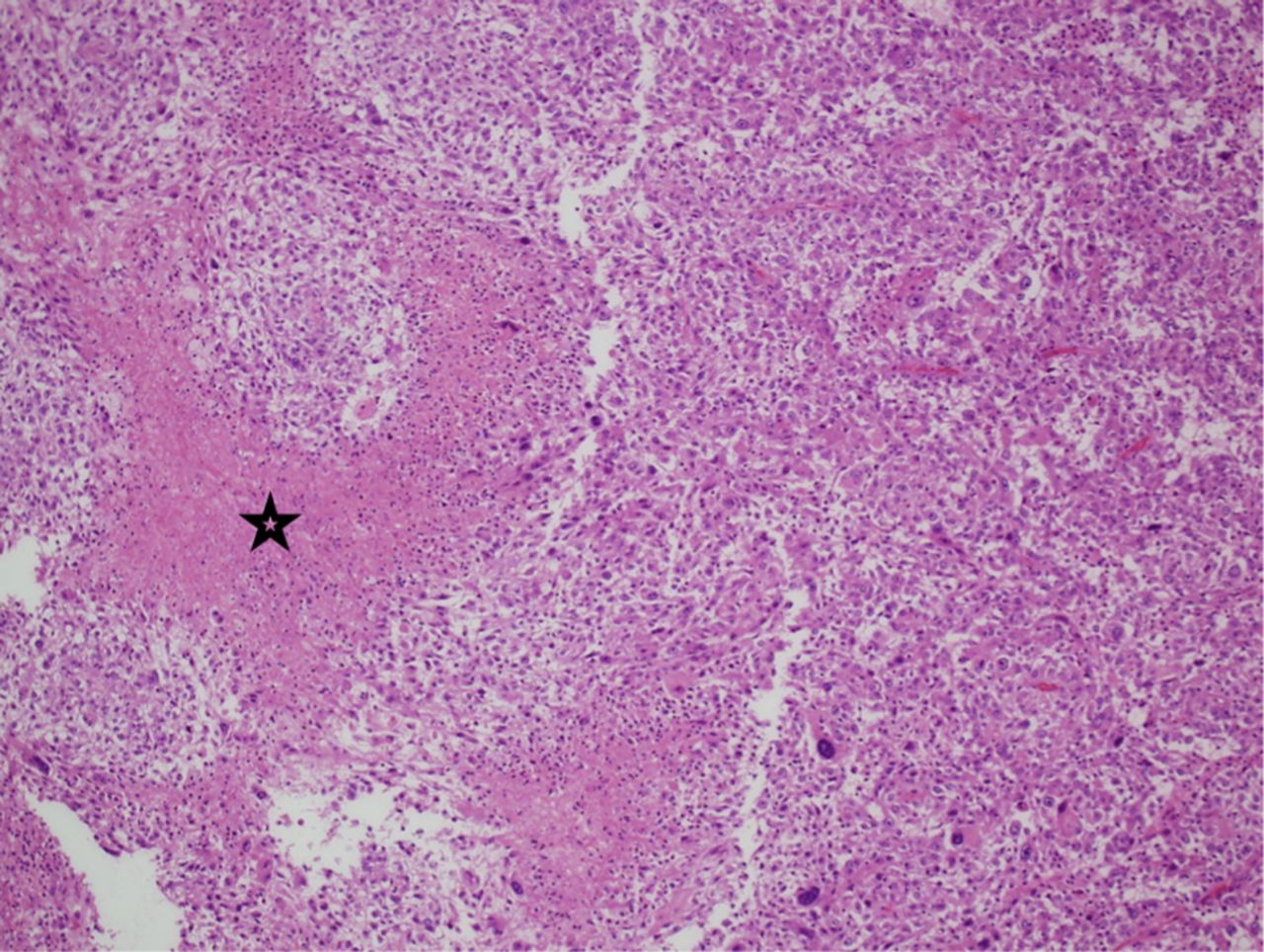
Pathological examination of the initially resected tumor revealed a relatively demarcated tumor composed of a combination of highly pleomorphic cells, bizarre giant cells (Figure 3A), rhabdoid-like cells (Figure 3C), and focally mononuclear spindled cells (Figures 3A & 3B). Small foci of conventional SEGA are seen at the periphery of the tumor (Figure 3D). There is only focal expression of glial fibrillary acidic protein and synaptophysin. Neurofilament protein shows a solid, non-infiltrative growth pattern devoid of axons for most of the part. Expression of thyroid transcription factor-1 was focal. There was focal increase in mitoses including atypical forms; the MIB-1 proliferation index in these areas was about 50%, and the p53 expression was diffused and strong. There was no microvascular proliferation or necrosis. Based on these findings and the history of TSC, the tumor was considered an atypical form of SEGA. The recurrent tumor showed predominance of smaller neoplastic cells. In addition, there were foci of necrosis and diffusely high MIB1 proliferation index of more than 60% (Figure 4).

Histopathological examination of the initial tumor with atypical features. A) Mild cellular pleomorphism (upper left) adjacent to more monomorphic and spindled glial cells (lower right). Note the bizarre atypical mitotic figure (arrow), (Hematoxylin and Eosin, x200). B) Bizarre tumor giant cells are seen at the lower right and mononuclear cells at the upper left. Two atypical mitotic figures are noted (arrows), (Hematoxylin and Eosin, x200). C) Rhabdoid-like tumor cells (Hematoxylin and Eosin, x200). These retained INI-1 expression (not shown). D) Tumor cells at the periphery has conventional morphology of SEGA (right) and appear well demarcated from adjacent CNS tissue (left), (Hematoxylin and Eosin, x100)
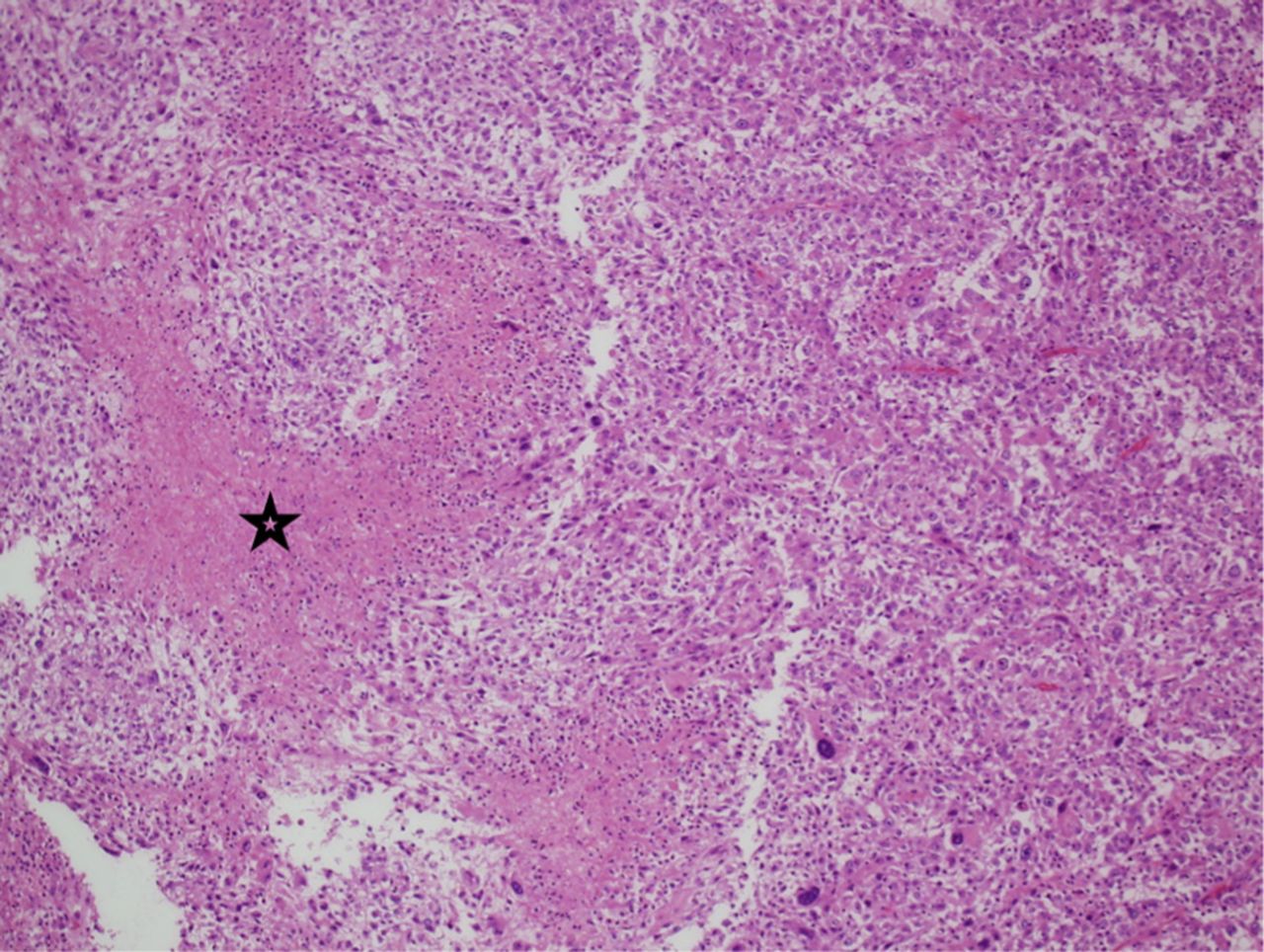
Histopathological examination of the recurrent tumor. The recurrent tumor is composed predominantly of small mononuclear cells Necrosis is depicted in this image (star), (Hematoxylin and Eosin, x40)
Follow up and outcome
Post operation, the patient improved clinically and was referred to Pediatric Oncology. He was started on procarbazine, lomustine and vincristine chemotherapy regimen. The patient was followed up for 5 years, and no radiological recurrence or clinical deterioration was observed.
Discussion
Pathological brain lesions in tuberous sclerosis include cortical tubers, subependymal nodules, SEGA and white matter abnormalities. The SEGAs occur in about 5-20% of TSC patients and can cause significant morbidity and mortality if left untreated. It is located exclusively near the foramen of Monroe causing obstructive hydrocephalus. They may rarely be present at other sites, such as the atrium of the lateral ventricles, the temporal horns, the fourth ventricle, and the third ventricle. The presence of SEGA at extra ventricular locations is extremely rare.3
The SEGAs have benign pathologic features, namely, slow growth, no brain edema, and minimal invasiveness.3 The average age of patients with SEGA is 9 years. The SEGAs are histologically benign and generally do not undergo malignant transformation. They are well circumscribed, often calcified tumors composed mainly of large, plump cells resembling gemistocytes and a spectrum of astroglial phenotypes. Typical appearances range from polygonal cells with abundant, glassy cytoplasm to smaller, more elongate elements within a variably fibrillar matrix.4 Proliferative index as measured by Ki-67 (MIB-1) is generally low (mean 1.5-7%), providing further evidence for the benign nature of these neoplasms. Though extremely uncommon, craniospinal dissemination has been reported in SEGA with increased MIB-1 without malignant features.5
Multiple factors have been identified in literature that are associated with more aggressive forms of SEGA, clinically or radiologically. Congenital SEGA and SEGAs in infancy are associated with higher growth rate than older age. They may have contiguous TSC2/ PKD1 deletion and resemble malignant glioamas pathologically.6-8 Our patient was only 11 months of age. Atypical SEGA occasionally show considerable increased mitotic activity, endothelial proliferation or necrosis. Grajkowka et al6 reported three cases of SEGA with high-grade histopathological features including gemisocytic cells, perivascular pseudorosettes, necrosis, atypical mitotic figures, microvascular proliferation and Ki Index of 15-20%. Unfortunately, there was no long follow-up to determine the clinical course of these atypical forms. However, these features are not necessarily indicative of malignant progression.9 In our case, necrosis, abnormal mitosis and vascular proliferation were present in the recurrent tumor.
Proliferation index was extremely high (~50%), indicating malignant behavior. The patient was followed up for 5 years and there was no recurrence, which may be due to the adjuvant chemotherapy treatment. This treatment may have delayed its recurrence compared to the initial fast recurrence.
Atypical SEGA can be misdiagnosed as glioblastoma. De novo glioblastoma or malignant transformation of SEGA is extremely rare with only few cases published in literature so far.2 None of these cases confirmed transformation of SEGA into glioblastoma, but rather de novo formation of glioblastoma, or possible misdiagnosis, which can lead to unnecessary aggressive medical treatment.10 Other possible hypotheses are that these cases are atypical forms of SEGA rather than glioblastomas, similar to our reported case. This case is unique in many aspects. First, SEGA occurred at a very early age. Secondly, it was situated in an extra-ventricular location. Thirdly, it has atypical histopathological features and very high proliferation index. Fourthly, it recurred very quickly, which is unusual for SEGA. Lastly, it presented in infantile age, consistent with the few published reports in literature that younger age can be associated with more aggressive forms of SEGA and faster growth rate.
In conclusion, Infantile atypical SEGA may have aggressive clinical course, earlier recurrence and worse prognosis than ordinary forms of SEGA. Differentiating glioblastoma from ordinary SEGA or atypical aggressive forms of SEGA is challenging. We suggest that the cases with unique histopathological findings in this age group might be termed as atypical SEGA. Further clinical studies are needed to differentiate it from ordinary SEGAs and glioblastoma and to determine its long-term outcome and prognosis.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received August 12, 2019.
- Accepted September 15, 2019.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.