


Abstract
Patients with isolated neurosarcoidosis (NS) can present with neurological symptoms that mimic other neurologic conditions, such as multiple sclerosis (MS). In this article, we present a case of 25-year-old man with a transverse myelitis. Magnetic resonance imaging (MRI) of the brain and spine showed several, periventricular, infratentorial, and spinal cord white matter lesions. He was diagnosed with MS and was started on fingolimod, which did not result in any improvement. Follow-up brain and spine MRI showed a longitudinally extensive lesion with leptomeningeal enhancement. Leptomeningeal and cerebellar biopsy showed non-necrotizing granulomas consistent with neurosarcoidosis. Fingolimod was discontinued. The patient was treated with oral prednisone and infliximab, which ceased the progression of the disease and allowed for slow improvement. Incorrectly diagnosing NS with MS or vice-versa, not only deprives patients of beneficial therapy, but also potentially exposes them to therapies that may be harmful.
Sarcoidosis is a systemic, granulomatous inflammatory disorder with protean manifestations. It is estimated that less than 1% of patients with sarcoidosis have isolated central nervous system (CNS) involvement, without systemic evidence of the disorder. Patients with isolated neurosarcoidosis (NS) can present with neurological symptoms that mimic other neurologic conditions, such as multiple sclerosis (MS). Such symptoms, which can affect timely diagnosis, can present a real challenge to neurologists. Incorrectly diagnosing NS for MS, or vice versa, might not only deprive patients of beneficial therapy, but also potentially expose them to harm.
Case Report
Patient information
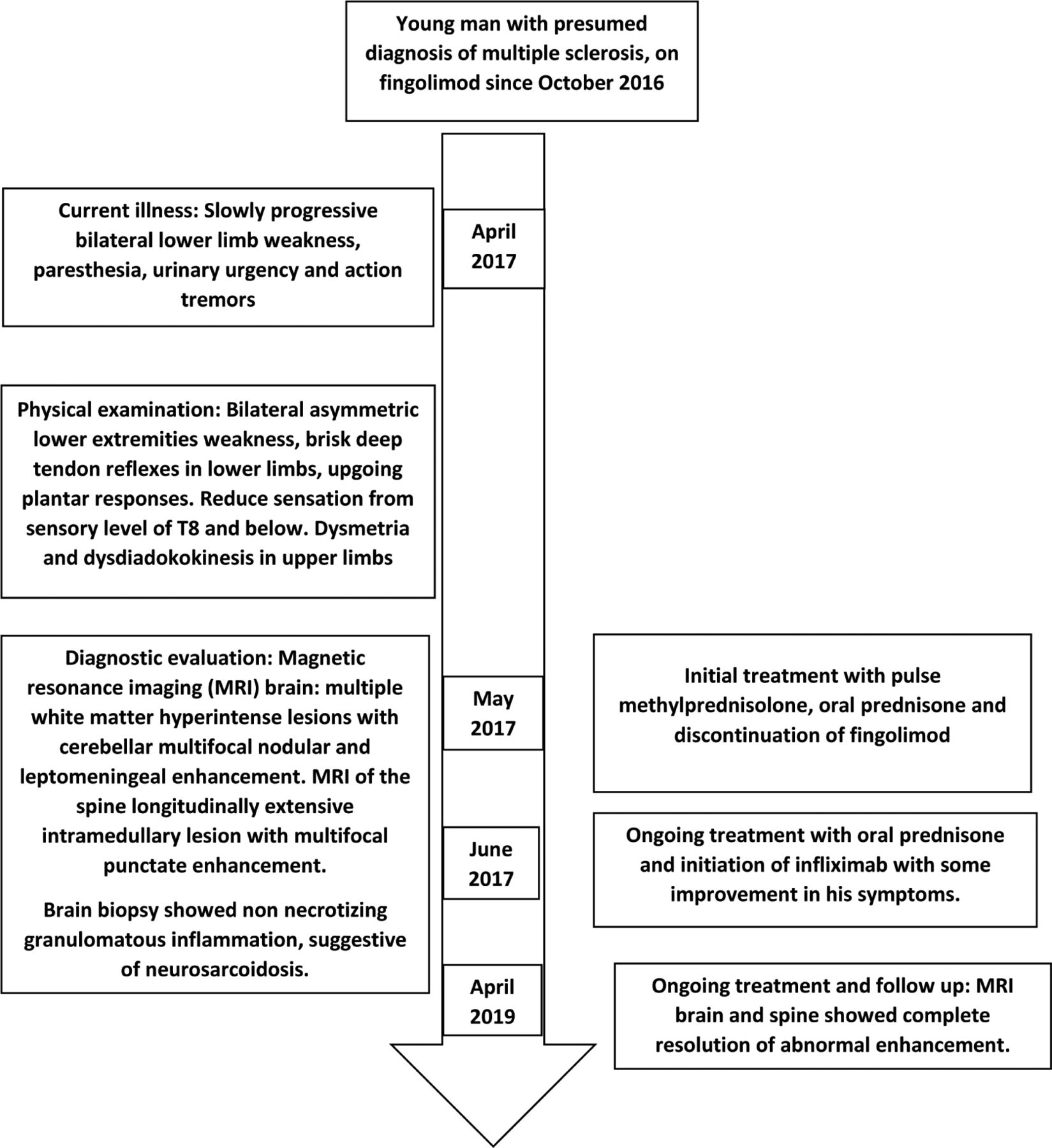
A 25-year-old man with a relapsing-remitting multiple sclerosis (RRMS) was diagnosed in October 2016, in another hospital. The patient had reported that during the previous one and a half years, he had had 2 episodes of lower limb weakness, numbness, and imbalance. He underwent intravenous methylprednisolone therapy, which resulted in partial recovery. The initial diagnosis of RRMS was made at another center, based on clinical and radiographic examination. He was prescribed 0.5 mg fingolimod once daily. After 6 months of treatment, he presented at our emergency department with progressive bilateral lower limb weakness, that had been ongoing for 4 weeks. He reported paresthesia of the right distal lower extremity and bilateral upper extremities, action tremors and urinary urgency. His symptoms were progressive, and he had difficulty moving without support, requiring a wheelchair for ambulation.
He had no history of headache, vision loss or other neurological symptoms. A comprehensive review found no evidence of fever, appetite changes, weight loss, cough, shortness of breath or chest pain. Family history was not significant. He was working as a soldier.
Clinical findings
The patient’s vital signs were within the normal range. Head, neck, chest, and cardiovascular examination were normal. He was alert with intact cognition. His language and cranial nerve examination were normal. Despite a spastic catch in his bilateral lower extremities, he had normal muscle bulk. Muscle power was 5/5 (Medical Research Council grade) throughout, except in the right hip flexors, knee flexion as well as ankle dorsiflexion, where it was 3/5. Power in the left hip flexors, knee flexion and ankle dorsiflexion were 4/5. Deep tendon reflexes were graded 3+ in the bilateral lower extremities and 2+ in the upper extremities. Plantar responses were upgoing, bilaterally. Sensation for pinprick, temperature and light touch was reduced from sensory level T8 downwards. Further, vibration and proprioceptive sensation were reduced in the distal lower extremities. Cerebellar examination disclosed dysmetria and dysdiadokokinesis in the bilateral upper extremities. Cerebellar examination could not be completed in the lower extremities due to weakness. Similarly, a gait examination could not be performed as the patient was unable to stand up without falling.
Diagnostic assessment
Magnetic resonance imaging (MRI) of the brain, with and without contrast, showed multiple non-enhancing subcortical and periventricular white matter hyperintense lesions. In addition, cerebellar multifocal tiny nodular and leptomeningeal enhancement was noted (Figure 1). The MRI of the spine showed a longitudinally extensive intramedullary lesion from T1-T6, with cord swelling and multifocal punctate enhancement (Figure 2). Cerebrospinal fluid (CSF) was clear and colorless, with 4 white blood cells/mm3 and normal glucose, protein levels and cytology. CSF oligoclonal bands were positive. Angiotensin-converting enzyme (ACE) levels in CSF and serum were normal. Blood and CSF cultures, were negative. The serum aquaporin 4 antibody test and autoantibodies associated with vasculitis were negative. Extensive investigations of malignancy were unremarkable, including a whole-body computed tomography scan, as well as a whole-body positron emission tomography scan. Given the neuroimaging abnormalities and the progressive course of neurological symptoms, the decision to conduct a leptomeningeal and cerebellar biopsy via suboccipital craniotomy was made. The biopsy results confirmed non-necrotizing granulomatous inflammation, suggestive of NS. Histochemical stains were negative for acid-fast bacilli and fungal elements. There was no evidence of malignancy.
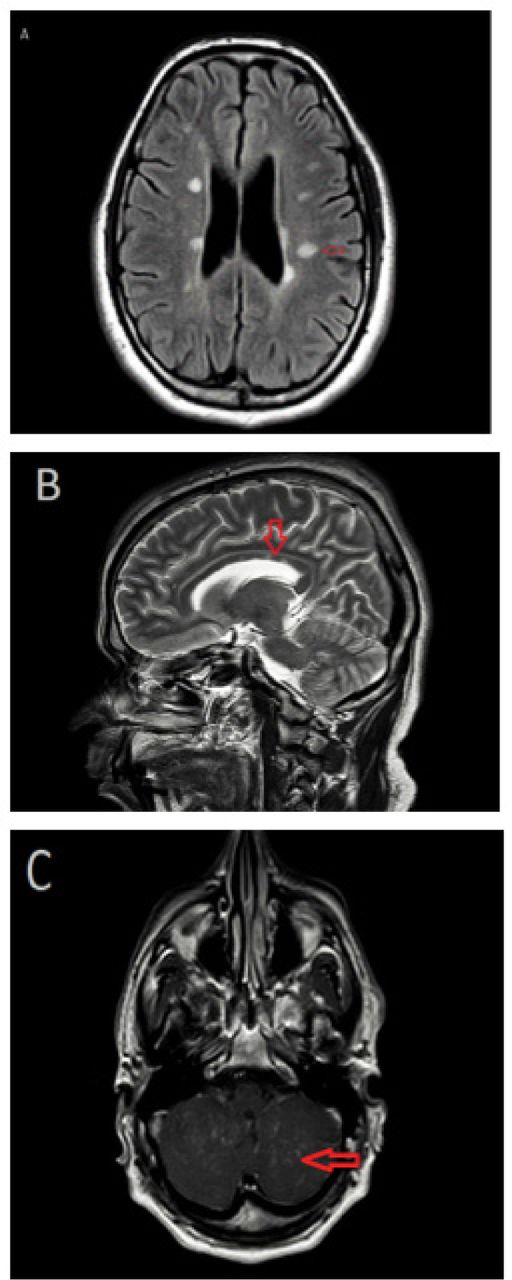
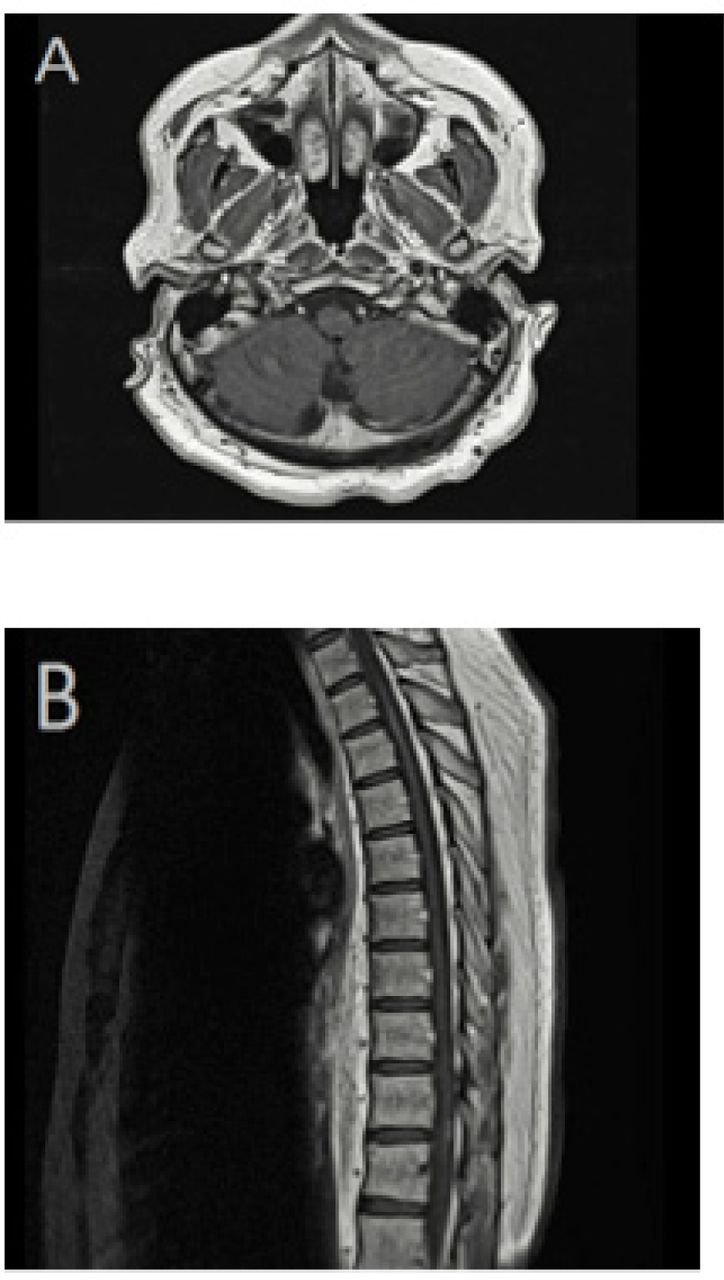
Magnetic resonance imaging (MRI) of the brain with and without contrast. A) Axial fluid-attenuated inversion recovery (FLAIR) MRI image demonstrating periventricular and subcortical white matter lesions. B) Sagittal T2-weighted image showing corpus callosal white matter lesions. C) Axial T1-weighted image with contrast highlighting cerebellar leptomeningeal enhancement.
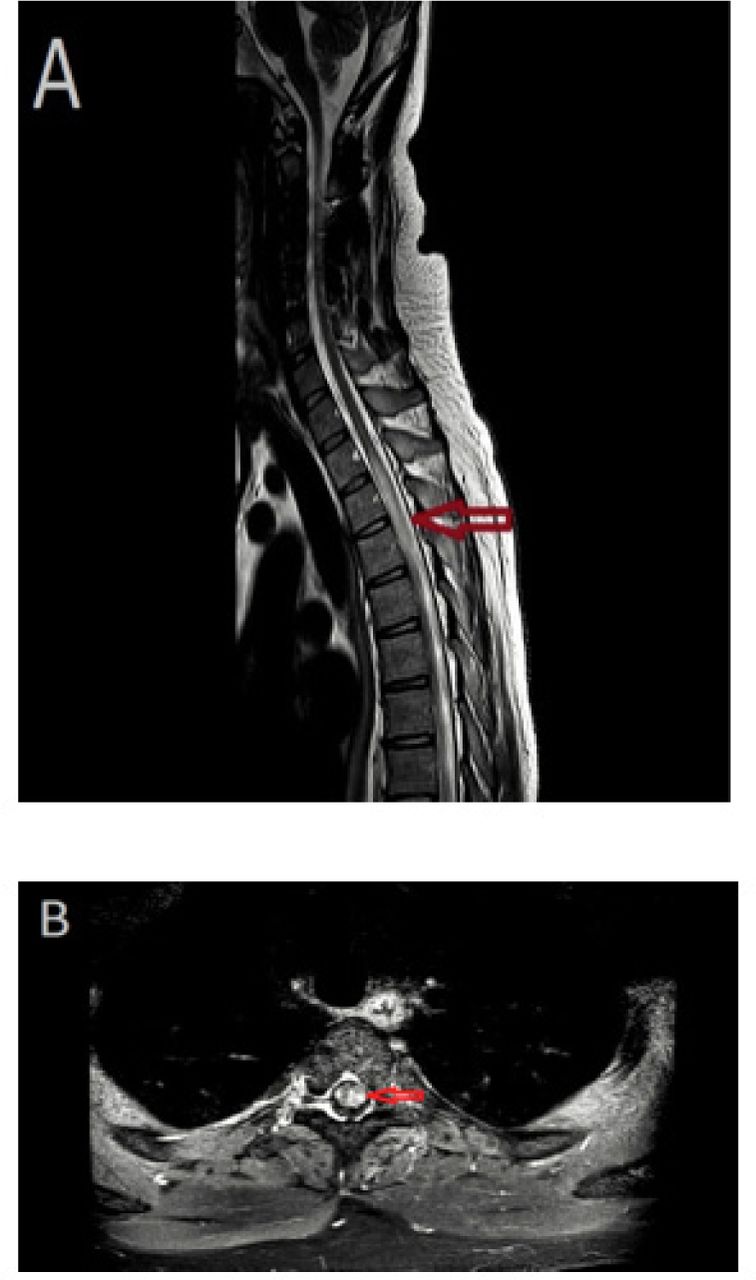
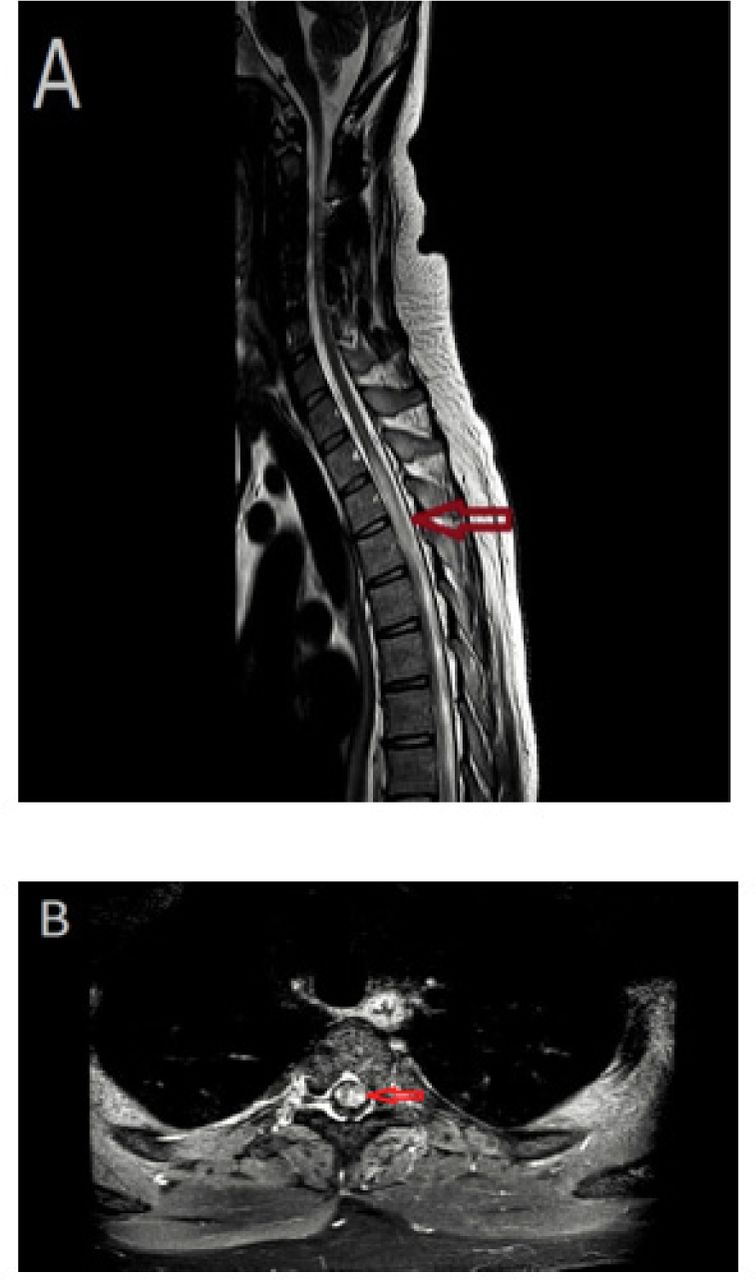
Magnetic resonance imaging (MRI) of the spine with and without contrast. A) T2 sagittal image demonstrating a longitudinally extensive lesion from T1-T6, with cord swelling. B) Axial T1-weighted image, with contrast, showing multifocal punctate enhancement.
Therapeutic intervention
Based on the neuroimaging abnormalities, fingolimod was discontinued. He was treated with pulse methylprednisolone therapy, followed by tapering oral prednisolone. After reviewing the biopsy result, the diagnosis of neurosarcoidosis was confirmed and the patient was started on Infliximab (5 mg/kg on Weeks 0, 2, 6 and 8). This stopped the disease progression and allowed for slow improvement.
Follow-up and outcomes
Following the initiation of infliximab treatment, the prednisone dose was reduced gradually to a maintenance dose of 5 mg daily, with no further relapses or disease progression. The last brain and spine MRI in April 2019 showed a complete resolution of the cerebellar leptomeningeal enhancement and intrinsic cord signal abnormalities (Figure 3). The patient required intense physiotherapy
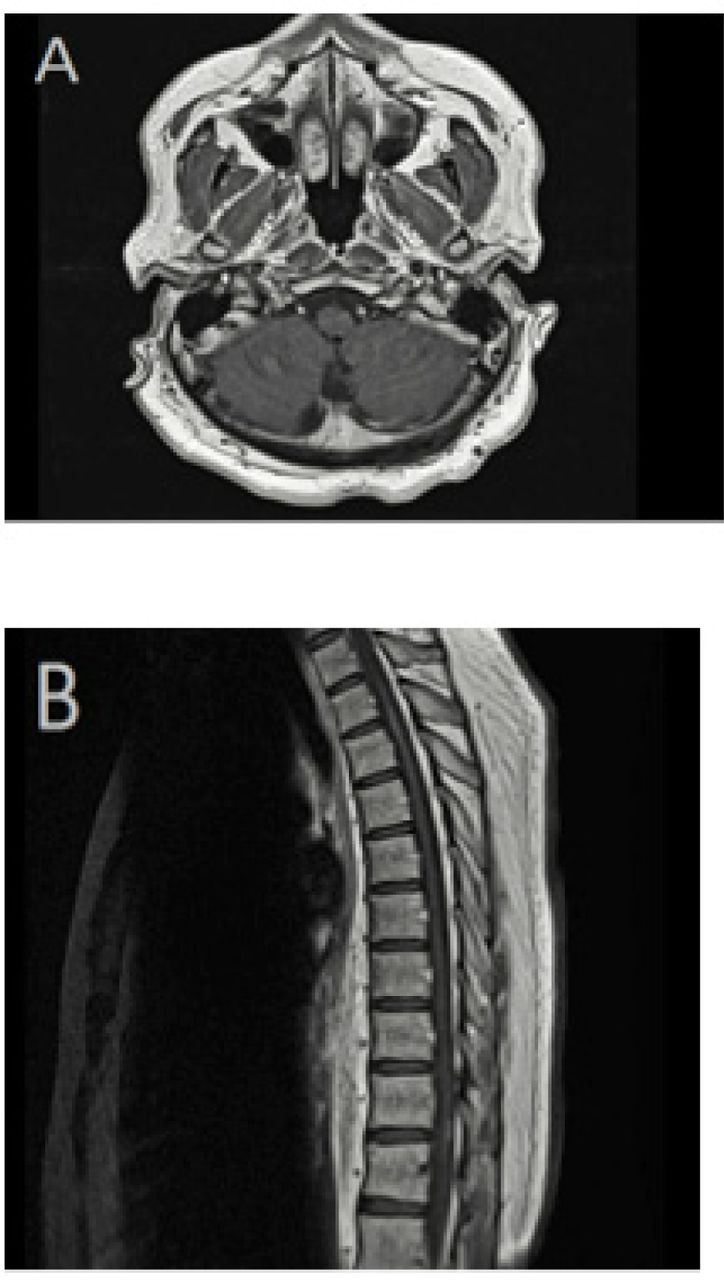
Magnetic resonance imaging (MRI) of the brain and spine with contrast. A) Axial T1-weighted image, with contrast, showing complete resolution of cerebellar leptomeningeal enhancement. B) Sagittal T1-weighted MRI demonstrating an improvement of spinal cord abnormal enhancement.
Discussion
The incidence of sarcoidosis ranges between 1 and 70 per 100,000 persons-years worldwide, although it is dependent on the population’s ethnic background. 1 NS, where the nervous system is involved, is reported in 4-16% of patients with sarcoidosis.1,2 Nevertheless, NS is found in up to 25% of autopsies, suggesting a high percentage of asymptomatic, missed or misdiagnosed cases.3
Neurological symptoms may present initially or may develop after many years of a sarcoidosis diagnosis. A long-term study of 345 patients with sarcoidosis found that of the 3% (11 patients) with neurological involvement, cranial neuropathy was the most common neurological manifestation (45%, 5 patients), followed by peripheral neuropathy (27%, 3 patients) and meningitis (also 27%, 3 patients).1 The cranial neuropathy in NS most frequently affects cranial nerves VII, II and III.4 Patients may also present with any one of a wide variety of clinical features, such as facial palsies, optic neuritis, myelopathy, weakness of extremities, dystonic spasms, paresthesia and sensory abnormalities. Most of these symptoms are non-specific and can also be found in MS.2 Early-stage MRI findings in NS and MS may also be undistinguishable; thus, the misdiagnosis of NS for MS is often reported.

Timeline of the presented case.
A frequent MRI abnormalities in NS are multiple non-enhancing periventricular white matter lesions, which may mimic demyelinating disorders such as MS.4 A cohort study of head and neck MRIs, from 85 patients with NS, found leptomeningeal enhancement (the most common finding) in 35% of the patients as well as white matter and spinal cord involvement (30% and 23%, respectively).5 Conversely, in MS, no meningeal involvement should be present.4 On MRI, leptomeningeal enhancement is seen as a diffuse or focal gyriform or serpentine enhancement of the pial arachnoid and the adjacent dura mater.
In MS, MRI findings typically reveal T2-hyperintense lesions that are asymmetric and located in four topographies of the CNS: periventricular, cortical or juxtacortical, infratentorial and spinal cord.4,6 Although the focal white matter lesions seen in NS may easily be mistaken for the T2-hyperintense ovoid plaques that are characteristic of MS, their morphology (as well as the fore mentioned locations) can help with identification. Specifically, MS lesions have regular and distinct borders, while alternative diagnoses should be considered if the lesions have fuzzy and indistinct borders or are large and confluent.4
Longitudinally extensive transverse myelitis (LETM), an extensive involvement of the spinal cord, with at least 3 vertebrae in length, and more than half of the cord area affected, including the gray matter, may also be present in NS. Conversely in MS, spinal cord involvement of more than 3 vertebrae should raise a red flag, as spinal cord lesions in MS are short (<50% of cord area and peripheral).4 The LETM is most frequently associated with neuromyelitis optica.7 Another distinguishing MRI factor may be grey matter atrophy6 and the associated decrease in brain volume, especially neocortical volume (detected with T1 signal), which is already detectable in the early stages of MS.
Extra-neurologic presentation of sarcoidosis was found in all, but 1, of the 11 patients with NS in the study by Ungprasert.1 Importantly, the lungs were, by far, the most commonly affected extra-neurologic site (73%).1 A second study investigating 69 patients with NS found a similar proportion (71%) with abnormal findings on chest computed tomography (CT).8 Furthermore, an extensive review reported a wide range in the proportion of patients with NS with lung-involvement, between 31-86%.2 As numerous NS cases are misdiagnosed as MS, they are later correctly diagnosed on the basis of chest CT findings and confirmed with hilar node biopsy (positive for non-caseating granulomas in 57%).8,9 This evidence emphasizes the importance of thoracic imaging for the differential diagnosis of MS from NS. However, as in the case described in this report, an absence of pulmonary lesions does not necessarily rule out NS.
Another sensitive test for MS is the detection of intrathecal oligoclonal IgG from the (CSF), seen in 95-98% of patients. Nevertheless, it is important to keep in mind that oligoclonal bands (OCBs) are not specific to MS. CSF from patients with NS may also give positive OCBs, although it is rare, with studies reporting varied proportions between 3% to 22% of patients.5
The CSF lymphocytosis is another common finding in NS (64% of patients), as well as elevated CSF protein levels (62% of patients), except in peripheral neuropathy.1 Furthermore, patients with diffuse leptomeningeal enhancement on MRI have been found to be associated with significantly higher CSF white blood cell counts, CSF protein, and CSF lactate, but lower CSF glucose levels than patients without leptomeningeal enhancement. Elevated white blood cells and protein in the CSF are rare in MS.5
Another distinguishing factor obtained from the analyses of CSF is the increased level of angiotensin converting enzyme (ACE), but decreased ACE,2 associated with MS. In contrast, in NS, CSF ACE levels are usually normal, but serum ACE level may be increased.5 Finally, an interesting study aimed at distinguishing NS from MS discovered that soluble interleukin-2 receptor (sIL2-R) level in the CSF could be used to identify patients with NS with an overall accuracy of 93% compared to other CNS disorders including MS, bacterial and viral meningitis, and CNS vasculitis. Furthermore, this increase in sIL2-R in the CSF preceded the outbreak of new neurological symptoms and could prove helpful on initial presentation. Unfortunately, this test was not available for our patient.
Corticosteroids are the first-line treatment for NS, followed by steroid-sparing immune-modulating agents, if prednisone is insufficient.3 Anti-Tumor Necrosis factor (TNF)-α therapies, such as infliximab, have also shown successful outcomes in numerous studies, improving symptoms and sustaining remission.2 Paradoxically, an important complication of anti-TNF-α medications has been the new onset of a sarcoidosis-like reactions in patients receiving anti-TNF-α therapies for other conditions, namely Crohn’s disease.2,10 Given their immune modulatory mechanisms, infliximab and lenercept were used in patients with MS in clinical trials with remarkably unfavorable effects. Specifically, they have been associated with an increase in relapse frequency, and disability, suggesting that TNF-α may be implicated in CNS demyelination.10 These trials advised that anti- TNF-α therapy is harmful in MS patients.10,11 Therefore it was crucial that we rule out the diagnosis of MS in our patient and confirm a diagnosis of NS with the biopsy, before initiating infliximab therapy.
A few cases have been reported in which sarcoidosis developed after several years of using interferon-beta for MS patients. However, due to the rarity of these types of cases, it is not clear whether interferon-beta therapy can trigger sarcoidosis, as it may be a case of NS that was initially misdiagnosed as MS or even a case of NS with underlying MS, which have been occasionally reported.
In summary, a multidisciplinary approach is needed to diagnose NS correctly, as findings from any single imaging or clinical test are often non-specific. Further, the disease can manifest anywhere along the neural axis, producing a wide variety of unspecific symptoms, often resulting in misdiagnosis as MS upon initial presentation, as with our case,5,9 Recommendations from the Neurosarcoidosis Consortium Consensus Group and the World Association of Sarcoidosis and Other Granulomatous Diseases stress the need to rule out alternate causal factors (including infection and malignant neoplasm) in patients with symptoms suggestive of NS. This is especially important in light of evidence supporting the harmful effects of a NS therapy (anti-TNF-α) for MS,10 and vice versa, such as a MS medication (β-interferon) that has been linked to the development of sarcoidosis. Also emphasized by the Neurosarcoidosis Consortium Consensus Group, is the need for histological evidence by means of biopsy, whenever feasible, of non-neural tissue (for example, skin, lung) to identify systemic sarcoidosis or neural tissue to confirm NS with certainty. This supports our decision to perform leptomeningeal and cerebellar biopsies in our patient, as it was the only way to confirm a NS diagnosis and confidently commence treatment with anti-TNFα therapy.
Acknowledgement
We would like to thank Editage (www.editage.com) for English language editing. We would like to thank Dr. Muhammad Ejaz Ahmed for reviewing the radiological findings for clarity and correctness.
- Received May 30, 2020.
- Accepted July 15, 2020.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.