


Abstract
Rosette-forming glioneural tumors (RGNTs) are rare. Here, we report a case of RGNT of the fourth ventricle in an 18-year-old female. The patient presented with a 4-month history of headache and dizziness. Neurological examination showed papilledema, impaired tandem gait, and right-sided dysmetria. Radiological images showed a posterior fossa lesion in the fourth ventricle with hydrocephalus. An emergent ventriculostomy was performed followed by gross total surgical resection of the lesion. Histopathological examination confirmed the diagnosis of RGNT. The patient developed posterior fossa syndrome postoperatively which improved on follow-up. Although rare, RGNT should be considered in the differential diagnoses of posterior fossa lesions in young patients. Given its benign course, surgical resection remains the treatment of choice.
Rosette-forming glioneural tumors (RGNTs) are benign, rare, and unique central nervous system (CNS) tumors. Histologically, a RGNT is characterized by a glial component and a neurocytic component forming neurocytic rosettes and/or perivascular pseudorosettes.1 First thought to arise in the fourth ventricle, RGNTs are increasingly reported in other locations, including the pineal gland, hypothalamus, cerebellum, and spine. The contemporary literature on RGNTs is limited to case reports and case series focusing primarily on pathological descriptions with short follow-up periods. Although literature is growing on RGNTs, pertinent reports discussing the clinical course and outcomes are still lacking. In this article, we report an additional case of RGNT of the fourth ventricle and review the clinical presentation, diagnosis, radiological and pathological features, treatment, and outcomes.
Case Report
Clinical findings
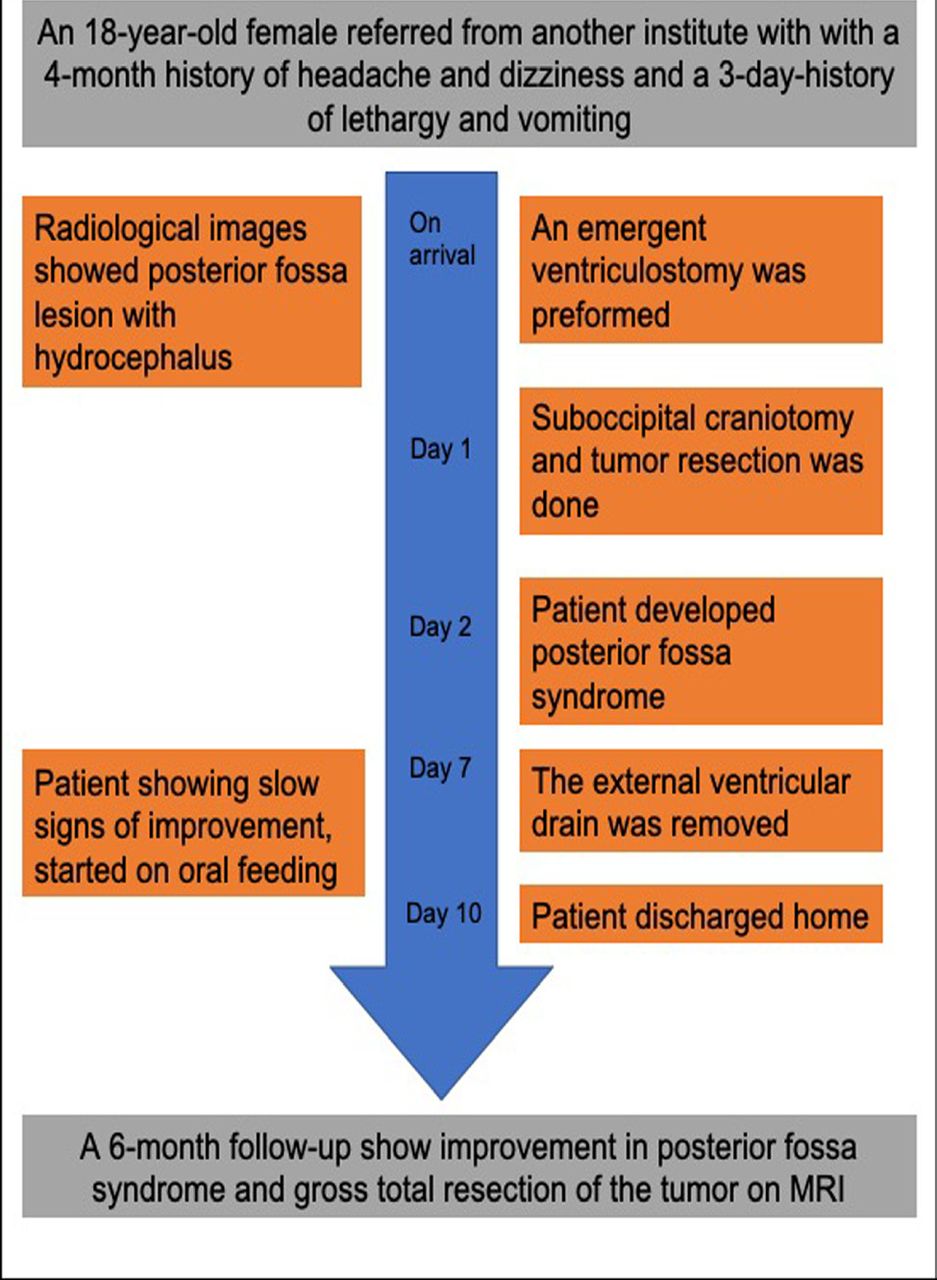
An 18-year-old female was referred to us from an outside institute with a 4-month history of headache and dizziness and a 3-day-history of lethargy and vomiting. A baseline neurological exam showed papilledema, impaired tandem gait, and right-sided dysmetria (Figure 1 shows the timeline of the patient presentation and clinical course).

- Timeline showing the clinical course of the patient and outcomes.
Diagnostic assessment
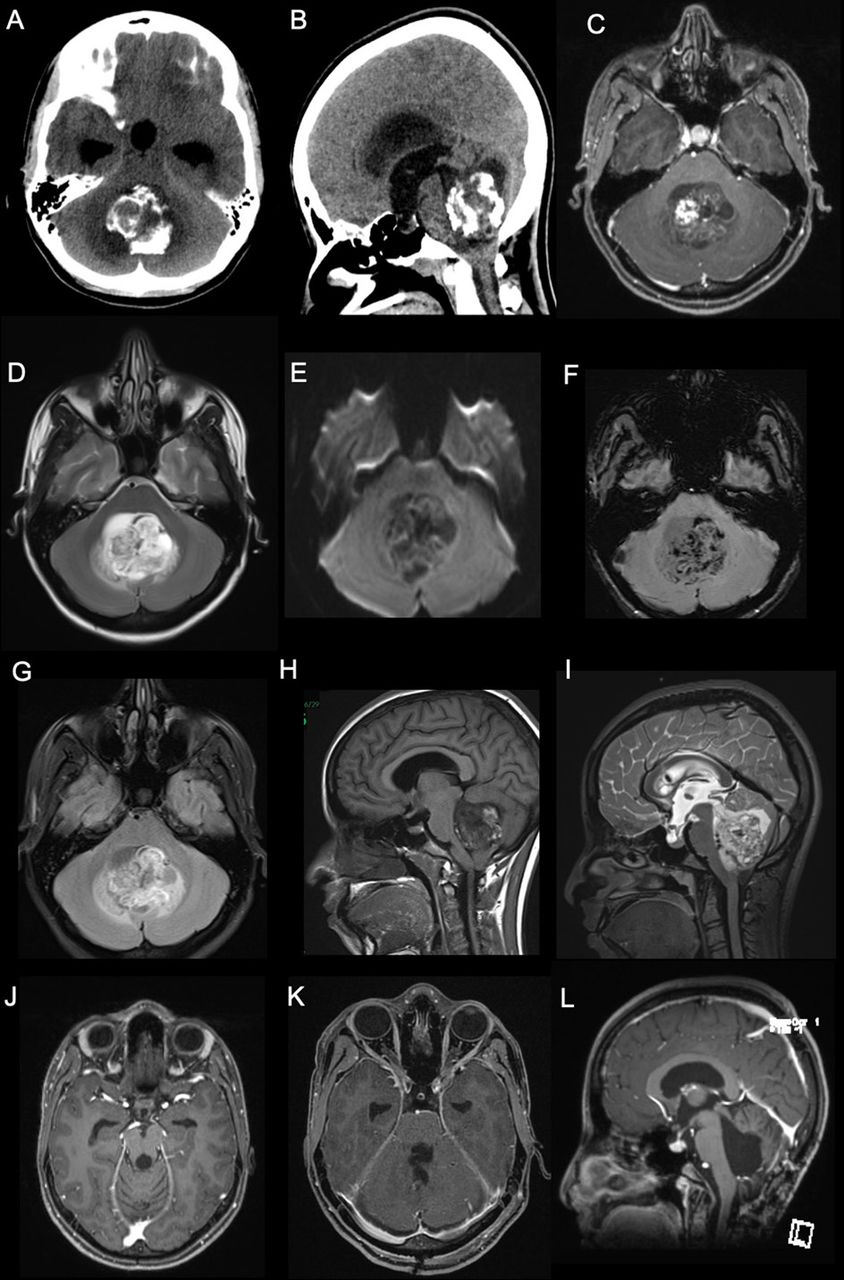
Radiological images showed a posterior fossa mass with coarse calcifications and a cystic component as well as hydrocephalus on computed tomography (CT) (Figure 2 A, B). Brain magnetic resonance imaging (MRI) showed a mass in the fourth ventricle with solid and cystic components. Both components were heterogenous, hypointense on T1, and hyperintense on T2. The lesion demonstrated heterogenous enhancement with gadolinium (Figure 2 C-F).

- Pre-operative images. A and B) posterior fossa mass with coarse calcifications and a cystic component as well as hydrocephalus demonstrated on CT scan. C and H) T1-weighted post contrast MRI showing a hypointense lesion with heterogenous enhancement with gadolinium. D and I) Axial and sagittal T2-weighted MRI demonstrating heterogenous, hyperintense lesion in the fourth ventricle. E) Diffusion weighted images showing a large heterogeneous cystic lesion centered within the fourth ventricle compressing the adjacent structures with no significant restricted diffusion. F) Susceptibility-weighted images showing multiple areas of susceptibility effects in the lesion corresponding to calcification with some areas representing hemorrhage. G) The lesion again seen demonstrating heterogeneous signal intensity with adjacent small edema in the cerebellum on Fluid attenuated inversion recovery images (FLAIR). J-L) Postoperative MRI obtained at 6-month postoperatively showing no evidence of recurrence.
Therapeutic intervention
As the patient was admitted to our hospital over the weekend with new symptoms of lethargy and drowsiness (symptoms of high intracranial pressure) and as she presented during the very early phase of the COVID-19 pandemic where the OR schedule was difficult to control, an external ventricular drain (EVD) was inserted on emergency basis. A suboccipital teleovelar gross total surgical resection was performed the following day.
The intra-operative squash preparation revealed neurocytic ill-defined rosettes surrounded by a loose fibrillary background (Figure 3A). The biphasic nature of the lesion was apparent in the frozen section. Histologic examination of the formalin-fixed and paraffin-embedded specimen revealed a rather discrete neoplasm with a biphasic pattern: one pattern was characterized by neurocytic rosettes (Figure 3B), and the other pattern resembled pilocytic astrocytoma, consisting of small and bland cells with oval nuclei and thin, hair-like processes. Numerous eosinophilic, red Rosenthal fibers were noted (Figure 3C). Microcalcifications were also observed. The rosetted neuronal cells showed clear cytoplasm, thus mimicking oligodendroglia (Figure 3D). Immunohistochemical staining for synaptophysin showed strong reactivity, particularly within the center of the rosettes (Figure 3E). Glial fibrillary acidic protein (GFAP) and S100 showed immunoreactivity in the pilocytic astrocytoma-like component (Figures 3F, G). No mitoses were noted, and the MIB-1 labeling index was very low throughout. The features were consistent with those of World Health Organization (WHO) Grade I RGNT. Vessels within the tumor were thickened with hyalinization (Figure 3H).

- Histopathological and immunohistochemical findings of rosette-forming glioneuronal tumors, A) A smear shows true rosettes embedded in a loose fibrillary background, B) Hematoxylin and eosin (H-E) staining shows a ring like neurocytic rosette arranged around a neuropil core, C) Pilocytic astrocytoma like component exhibiting rosenthal fibers, D) Oligodendrocyte-like cells with perinuclear halo. (E) Neurocytic rosettes are positive for synaptophysin, f, g) GFAP and S100 showing positive staining in the pilocytic astrocytoma area, H) Prominent hyalinization in the glial area.
Follow-up and outcomes
Gross total surgical resection was achieved. Postoperatively, the patient developed posterior fossa syndrome in speech and swallowing domains, but showed slow signs of improvement on follow-up. The external ventricular drain was kept for one week and eventually removed. MRI obtained at 6-month follow-up showed no evidence of recurrence (Figure 2 G-I) At the latest follow-up visit, the patient was able to ambulate independently with good gait control. She was tolerating orally with some residual speech difficulties
Discussion
RGNT was first described by Komori et al.2 in a series of 11 patients with lesions located in the fourth ventricle, cerebellar vermis, and cerebral aqueduct. Following their work and subsequent cases, it was included in the 2007 WHO classification as “a primary central nervous system tumor of the fourth ventricle” within the group of glioneuronal tumors.1 More cases were identified in other anatomical locations leading to a modification of its initial description in the 2016 WHO classification to “rosette-forming glioneuronal tumors.”3 RGNTs are considered benign WHO grade I primary mixed neuronal–glial tumors with favorable prognosis and outcomes.1-3
The etiology of RGNT has been studied by many authors over the past decade. RGNTs were speculated to be embryologically derived from the subependymal plate of the second germinal layer.2 Others proposed that they originate from cells in the cerebral/cerebellar internal granule layer with the capacity for both neuronal and glial differentiation.4 A periventricular stem cell origin with biphenotypic differentiation has also been suggested.5
RGNTs are more commonly observed in young adults with a mean age of 23 years and a slight female predominance. Headache, dizziness, visual disturbances, ataxia, tinnitus, nausea, and emesis are the most common clinical presentations of RGNT of the fourth ventricle. Incidentally discovered lesions have been also reported.6
While it was initially described in the fourth ventricle and cerebellum, RGNTs are increasingly reported in other locations, including the pineal gland, spinal cord, lateral ventricles, optic chiasm, hypothalamus, and pons. A recent systematic review identified a total of 26 cases of RGNT in the literature, and 10 of those were located in or extended to the fourth ventricle.6 Although we believe there are more cases in the literature, single reports discussing the clinical course and long-term outcomes of RGNT are still lacking.
Radiologically, RGNTs may show a cystic, solid, or a mixed cystic-solid pattern on MRI. In addition, these tumors are hypointense on T1-weighted MRI and hyperintense on T2-weighted MRI. The enhancement pattern with gadolinium is variable. Certain tumors will appear non-enhanced while others might appear heterogenous, rimmed, or with focal areas of enhancement. RGNTs typically show no evidence of diffusion restriction on diffusion-weighted imaging. Moreover, MR spectroscopy demonstrates an elevated choline value and reduced N-acetylaspartate value.7
Histologically, RGNTs are identified by biphasic mixed glial and neurocytic components. The glial component consists of Rosenthal fibers and eosinophilic granular bodies. The neurocytic component contains uniform neurocytic rosettes or perivascular pseudorosettes which may appear unanchored in microcytic mucinous areas. Moreover, mitosis and necrosis are typically lacking. Focal infarctions, microcalcifications, hemosiderin deposits, and vascular sclerosis may be identified. Immunohistochemically, positivity for synaptophysin, neuron-specific enolase, GFAP, and S100 are often observed.6
Given its rarity, the diagnosis of a RGNT may be challenging. Differential diagnoses of posterior fossa lesions in young adults include pilocytic astrocytoma, ependymoma, and choroid plexus papilloma. The presence of rosettes can help to differentiate RGNT from pilocytic astrocytoma morphologically. In addition, lack of dysembryoblastic neuroepithelial tumor (DNET) characteristic floating neurons can differentiate RGNT from DNET. Differential diagnoses of CNS tumors with rosette formation include ependymoma, astroblastoma, medulloblastoma, embryonal tumor with multilayered rosettes, pineoblastoma, and pituitary adenoma (Table 1).6
- Summary of CNS tumors with Rosette formation.
Surgical treatment remains the cornerstone in the management of RGNT with the aim of achieving gross total resection once possible. Nonetheless, no significant difference was found between gross total resection and subtotal resection for RGNT in regards to tumor progression.7 RGNTs are considered to be benign, with a low risk of recurrence. However, this view has been challenged by an increasing number of subsequent cases harboring the potential for malignant transformation and rapid progression.8 Pediatric patients, patients with solid RGNTs, and inadequate surgical resection (biopsy or partial resection) were identified as risk factors for tumor progression.7 The role of chemotherapy and radiation therapy as a primary or adjuvant treatment option is not yet established. Its implication for select aggressive RGNTs has been described.9
Posterior fossa syndrome refers to a constellation of transient mutism and emotional liability accompanied by motor, cognitive, and cranial nerve impairments following surgery to the posterior fossa. Although it is well-described in pediatric neurosurgery literature, the literature on posterior fossa syndrome in adults is sparse. Vermal splitting, deep cerebellar nuclear involvement, and disruption of efferent pathways in the superior cerebellar peduncle all contribute to the development of posterior fossa syndrome.10
Conclusions
Rosette-forming glioneural tumors are rare and unique CNS tumors. Although rare, RGNT should be considered in the differential diagnoses of posterior fossa lesions in young patients. Given its benign course, surgical resection remains the treatment of choice.
Acknowledgment
We would like to thank Dr. Abdulaziz Alsaad, Department of Radiology, King Fahad Medical City, for reviewing the figures.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received November 18, 2020.
- Accepted March 22, 2021.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.