


Abstract
15q11.2 BP1-BP2 microdeletion is related to clinical abnormalities including general developmental delay, speech and neuropsychiatric disorders, which is known as Angelman syndrome. However, the clinical penetrance and phenotype of 15q11.2 BP1-BP2 deletion is varied and confusing. Herein, we retrospectively described a 50-year-old male patient who manifested with progressive spastic paraplegia of lower limbs and episodic exacerbation. While brain MRI showed white matter hyperintensities, lacunes, cerebral microbleeds, enlarged perivascular spaces, and brain atrophy, mimicking small vessel disease. Next-generation sequencing combining multiplex ligation-dependent probe amplification identified a 253 kb 15q11.2 BP1-BP2 microdeletion, encompassing 4 conserved imprinted genes (NIPA1, NIPA2, CYFIP1 and TUBGCP5). This report will build new connections among spastic paraplegia, small vessel disease and 15q11.2 BP1-BP2 microdeletion.
15q11.2 BP1–BP2 (OMIM 615656) microdeletion refers to the deletion between breakpoint 1 (BP1) and breakpoint 2 (BP2) compassing 4 evolutionarily conserved nonimprinted genes (NIPA1, NIPA2, CYFIP1, and TUBGCP5) in the q-arm of chromosome 15. This deletion is clinically presented as Prader-Willi syndrome or Angelman syndrome (AS).1 In 2007, Murthy et al2 first reported AS in a 3.5-year-old boy due to a 253-kb microdeletion in 15q11.2 BP1-BP2, who presented with mental retardation, developmental delay, speech delay, and neurological disorders. Subsequently, the clinical spectrum of the 15q11.2 BP1-BP2 deletion has been expanded to neuropsychiatric disorders, autism spectrum disorders, epilepsy, general dysmorphic features, and so forth.2,3 But the presentation of progressive spastic paraplegia and small vessel disease has not been reported.
Cerebral small-vessel disease (CSVD) is a group of diseases based on brain imaging biomarkers, including recent small subcortical infarcts, white matter hyperintensities, lacunes, cerebral microbleeds, enlarged perivascular spaces, and brain atrophy.4 Patients present with recurrent stroke syndromes, cognitive impairments, gait dysfunction, and a general decline in function.5 Single-gene disorders are rare but an important cause for CSVD, including cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy caused by a mutation in the NOTCH3 gene, Fabry disease caused by the GLA gene, and so forth.5 This study aimed to report a case who presented with progressive spastic paraplegia, while the brain magnetic resonance imaging (MRI) revealed typical features of small-vessel disease. The genetic analysis identified 253-kb microdeletion in 15q11.2 BP1-BP2. The findings of this report may expand the phenotypic spectrum of 15q11.2 BP1-BP2 microdeletion.
Case Report
Patient information and diagnostic assessment
A 50-year-old male patient was admitted to Tongren Hospital, Shanghai Jiaotong University School of Medicine, with a 6-year history of progressive lower spasm and weakness. At 44 years of age, the patient experienced inflexibility and weakness of his left leg when he stepped on a bus. At 46 years of age, he had 2 episodes of exacerbation of left leg weakness and was treated for ischemic stroke but with no effect. Then, he had difficulty in climbing upstairs and riding a bicycle. At 47 and 48 years of age, he had 3 episodes of numbness and weakness of the left limbs. From this time, he developed urinary incontinence. At the age of 49 years, he had stiffness of the right leg. At admission when he was 50 years old, he came with the chief complaint of stiffness on the buttock and legs and difficult in walking.
The physical examination showed that the patient was conscious and speaking fluently. His muscle strength was MRC grade 4 on the left leg and normal on other extremities. He had brisk knee reflex (++++), positive ankle clonus, and positive Babinski sign on both lower extremities. His cognitive function was normal with the Minimal-Mental Status Examination score at 28/30 and Montreal Cognitive Assessment score at 27/30. He smoked (cigarette 20 pieces/day for 30 years), but he did not drink. Regarding the family history, his mother died of breast cancer at the age of 70 years, and his father died of ischemic stroke at the age of 76 years. His son, aged 26 years, was healthy. He reported no family history of a similar presentation.
Clinical findings
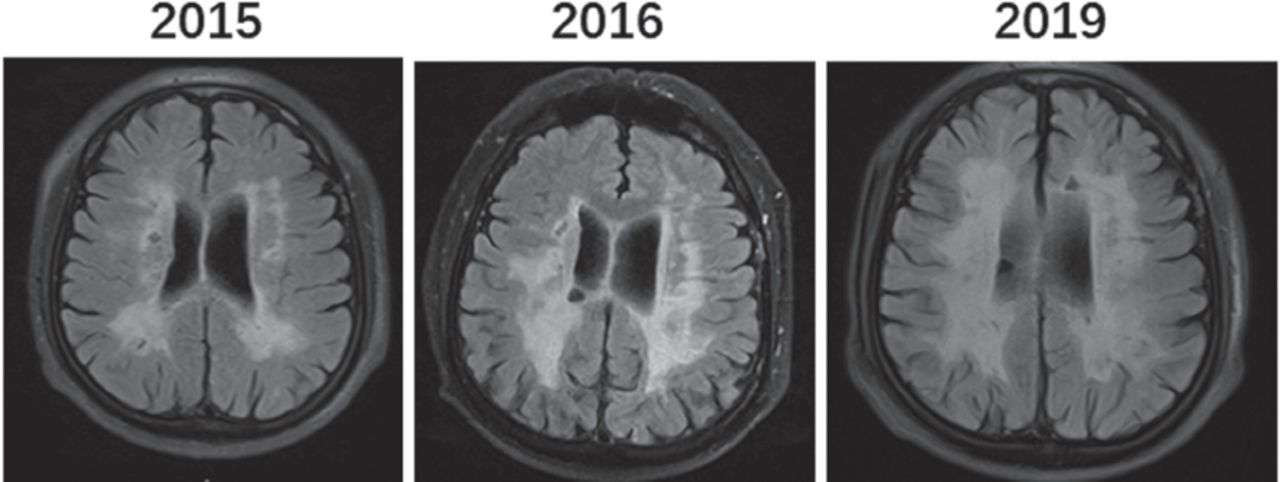
The regular blood tests including liver, renal, thyroid, and adrenal functions were all normal. His serum homocysteine level was mildly elevated at 24.3 μmol/L (normal range 4–15.4 µmol/L). He had elevated low-density lipoprotein level at 3.36 mmol/L (normal range <3.37 mmol/L). The lumbar puncture showed normal pressure, CSF protein level, and cell count, and he was negative for oligoclonal bands. Brain MRI showed diffusive white matter hyperintensities, lacunes, cerebral microbleeds, enlarged perivascular spaces, and cerebral atrophy (Figure 1A). His white matter lesions gradually enlarged with age from 2015 to 2019 (Figure 1C). Sagittal MRI of the spinal cord showed that the cervical and thoracic spines were normal (Figures 1B a-b). The diagnosis of small-vessel disease was based on the symptoms and signs of progressive spasticity of lower limbs and brain MRI findings of diffusive white matter hyperintensities, lacunes, cerebral microbleeds, enlarged perivascular spaces, and cerebral atrophy (Figure 1A and Figure 1C).
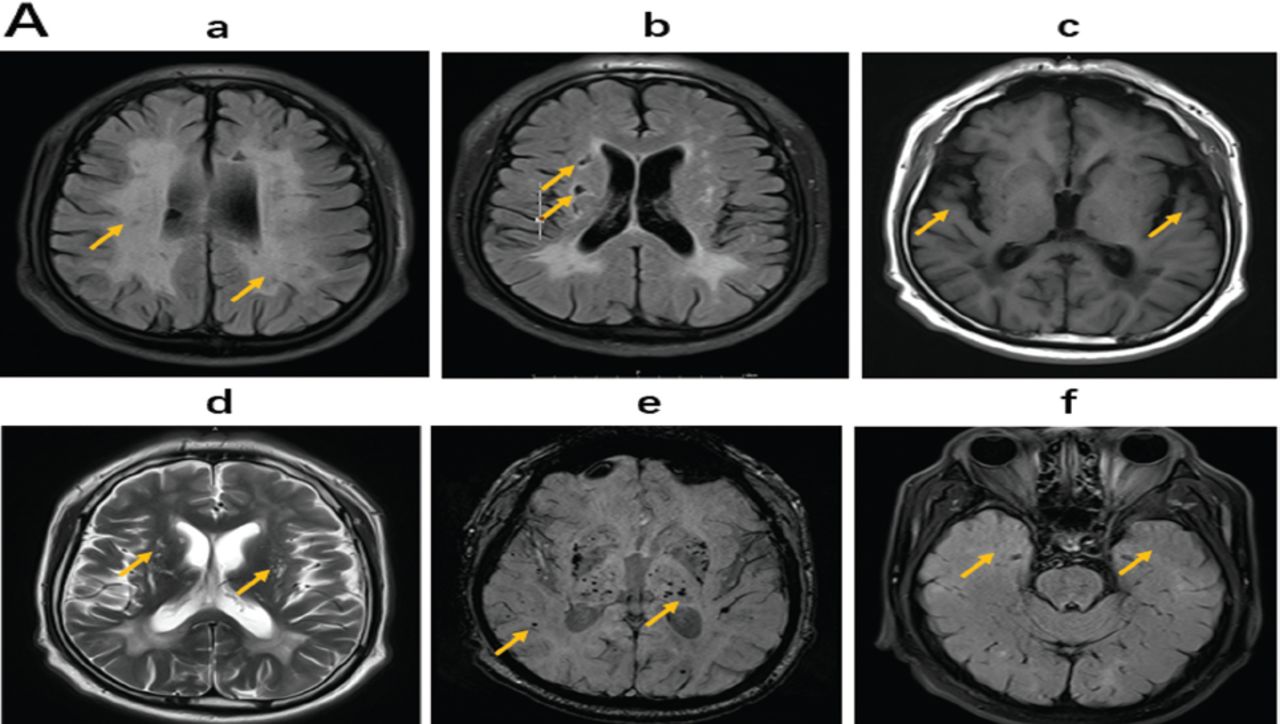
- Cranial MRI in 2019 at the admission Brain magnetic resonance imaging (MRI) indicated characteristics of cerebral small-vessel disease (CSVD) with (a, FLAIR) white matter hyperintensities, b, FLAIR) lacunes, c, T1) cerebral atrophy, d, T2) cerebral enlarged perivascular spaces, e, SWI) microbleeds in bilateral pons, basal ganglia, and subcortex. f) Bilateral temporal poles were intact in the temporal lobe.
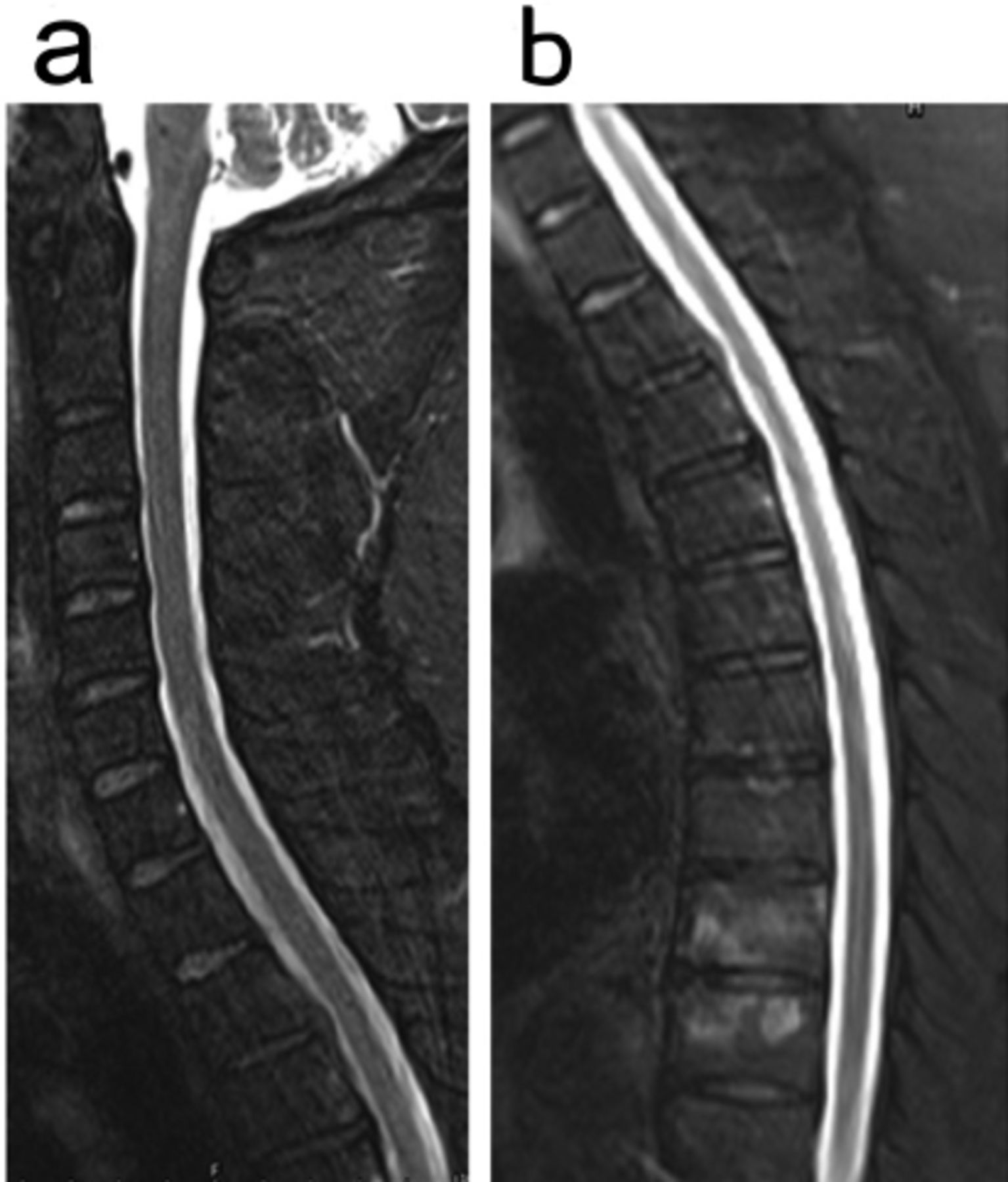
- Cranial MRI in 2019 at the admission brain magnetic resonance imaging (MRI) cervical and thoracis spinal cord scanning did not show spinal cord atrophy (a, b).

- Brain magnetic resonance imaging (MRI) (T2 FLAIR) indicated abnormal signals of periventricular white matter lesions gradual expanded from 2015 to 2019.
Through whole exome sequencing, a heterozygous 253-kb microdeletion was identified in the chromosome 15q11.2 (chr15: 22833477-23086385), which encompasses 4 nonimprinted genes (NIPA1, NIPA2, CYFIP1, and TUBGCP5) (Figure 2A). This 15q11.2 BP1-BP2 microdeletion was verified by multiplex ligation−dependent probe amplification (MLPA) according to the manufacturer’s instructions using the SALSA MLPA kit (MRC-Holland) (Figure 2B). Deep genetic evaluations were conducted to exclude HSP, metachromatic leukodystrophy, adrenoleukodystrophy, adrenomyeloneuropathy, metachromatic leukodystrophy, and Krabbe’s disease. Also, small-vessel diseases related to single genes including NOTCH3, HTRA1, GLA, and COL4A1–COL4A2. and so forth.
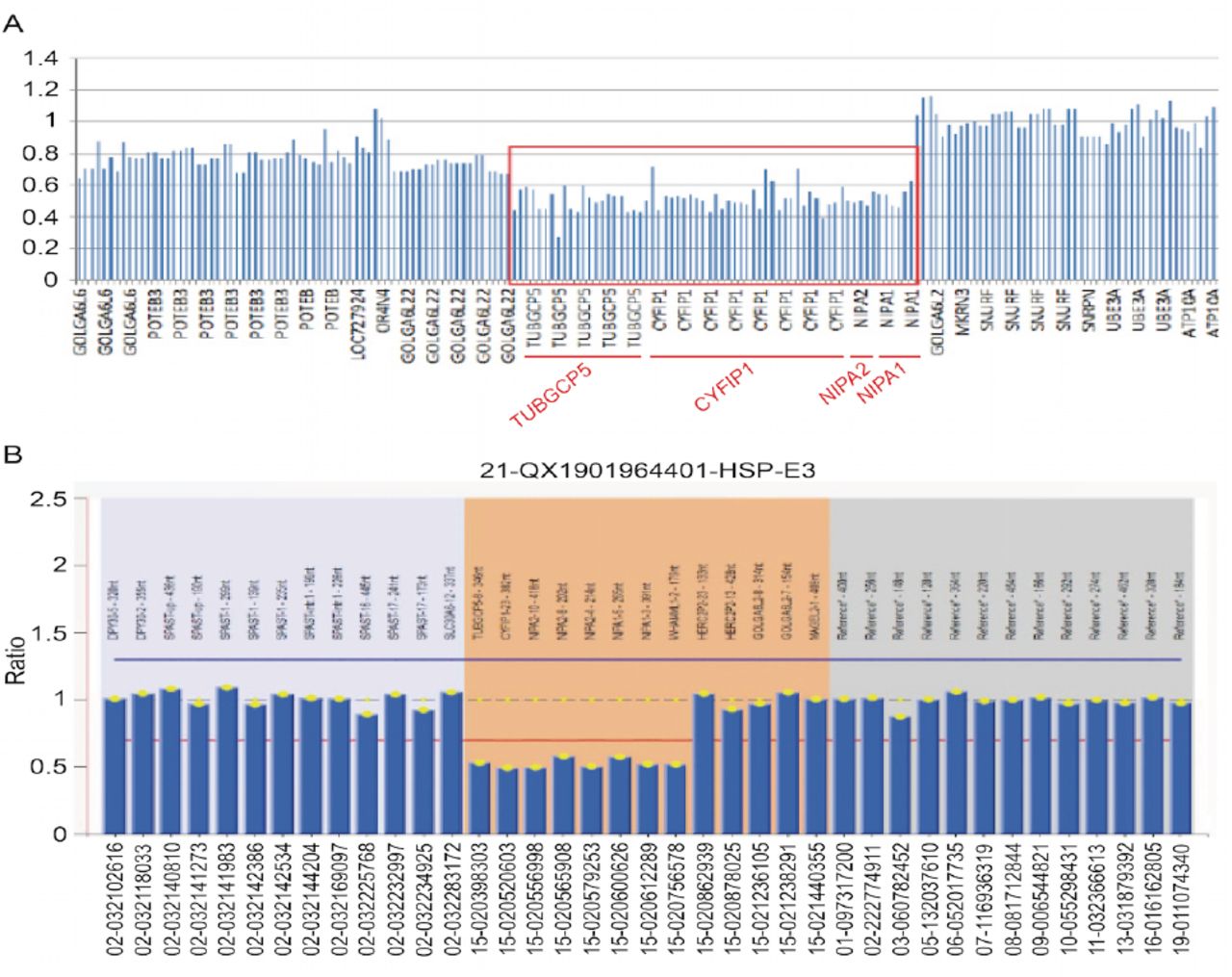
- NGS results showed heterozygous deletion variation on chr15: 22833477-23086385, encompassing 4 genes NIPA1, NIPA2, CYFIP1, and TUBGCP5 (A). This deletion was confirmed by MLPA (B).
Therapeutic intervention
He had a medical history of hypertension, which was properly controlled with nifedipine sustained-release tablets 30 mg/day and valsartan 100 mg/day. At 47 and 48 years of age, when he had numbness and weakness of the left limbs, he has been treated as multiple sclerosis using methylprednisolone 500 mg qd × 5 days, with limited alleviation. He had hyperlipoidemia and discontinued rosuvastatin 1 year ago. At this admission, the patient was treated with intramuscular injection of cobalamin 500 µg/day and oral folate 5 mg/day for a month, oral aspirin 100 mg/day, rosuvastatin 10 mg/day, and medicines for hypertension as usual, but showed no improvement.
Follow-up and outcomes
After discharge, we continued follow up the patient for 2 and a half years by the outpatient, with 6 months interval. He was keeping with the treatment of anti-hypertension medicines, oral aspirin 100 mg/day, rosuvastatin 10 mg/day. But his symptoms worsen gradually and he has to stay in wheelchair now (Table 1).
- Patient timeline of disease evolution.
Discussion
The 15q11.2 BP1–BP2 microdeletion syndrome has a variable phenotype, with low clinical penetrance. A retrospective analysis of 200 patients with 15q11.2 deletion found that developmental delay and speech delay accounted for 73% and 67% of cases, respectively.2 More than half of the patients suffered from writing (60%) and reading (57%) difficulties and memory problems (60%). Another report showed that 38.7% (31/52) of 15q11.2 microdeletion carriers had abnormal brain MRI cerebral atrophy, ventriculomegaly, cerebellar hypoplasia, and so forth.3 However, neither typical spastic paraplegia nor small-vessel disease has been reported due to this microdeletion. This study aimed to report a case of a patient carrying 253- kb microdeletion, who presented with progressive spastic paraplegia and features of small-vessel disease on brain MRI.
The CSVD is a group of diseases sharing similar brain MRI features but with diverse reasons and genetic backgrounds. Other than certain single gene mutations, multiple genes or genetic loci also contribute to the background of CSVD, which remain to be elucidated.5 Recently, new techniques have been used to identify common genetic variants associated with CSVD and MRI-defined covert CSVD properties. More than 50 independent genetic loci associated with the risk of CSVD have been identified through genome-wide association studies and transcriptome-wide association studies (TWAS).6 Among these, variants in or close to the COL4A1–COL4A2 loci (responsible for Type IV collagen mutation–related CSVD) and HTRA1 loci are related to the risk of CSVD. Kuuluvainen et al7 found that a 2.2-Mb interstitial 13q33.3q34 duplication encompassing the whole gene of COL4A1 and exons 1–4 of the COL4A2 gene was associated with CSVD with recurrent early-onset ischemic strokes.
The microdeletion in 15q11.2 BP1–BP2 related to small-vessel disease has not been reported. However, a report showed that variants on 15q22 nearest to MTFMT, SLC51B, and RASL12 were related to the white matter hyperintensity of CSVD.8 This 15q11.2 microdeletion reported here included four nonimprinted genes (NIPA1, NIPA2, CYFIP1, and TUBGCP5), all of which were expressed in the central nervous system and might play key roles in the development and functional maintenance of the brain. The NIPA1 gene encodes a magnesium transporter implicated in neuronal development and maintenance. The point mutations in the NIPA1 gene have been identified as disease causing hereditary spastic paraplegia type 6, which is characterized by the progressive spasticity of the lower limbs. The NIPA2 gene encodes a highly selective magnesium transporter. Missense mutations or insertions of NIPA2 may lead to childhood absence epilepsy. The CYFIP1 regulates the normal function of the Fragile X mental retardation 1 (FMR1) protein, and the mutation in the FMR1 gene is responsible for Fragile X syndrome. The TUBGCP5 is related to microtubules and involved in neurobehavioral disorders.9 The phenotype of spastic paraplegia and small-vessel disease on MRI does not mimic any clinical patterns of the 4 gene deficiencies separately. However, it has been reported that duplication on 15q11.2 encompassing these 5 genes (TUBGCP5, CYFIP1, NIPA2, NIPA1, and WHAMML1) contributes to Alzheimer’s disease.10
This study had some limitations. The patient’s parents have passed away and his son has not yet reached the age of onset; hence, the family co-segregation was not performed. We performed deep analysis on WES data to exclude possible mutations for progressive spastic paraplegia and known single-gene mutations for CSVD, but new mutations were possible. We still cannot totally rule out the possibility of CSVD caused by hypertension and smoking, concomitantly carrying a 15q11.2 BP1-BP2 microdeletion. In summary, we reported a case of a patient carrying 15q11.2 BP1–BP2 microdeletion who presented with spastic paraplegia and MRI features of small-vessel disease. The findings of this report might expand the phenotypic spectrum of 15q11.2 BP1-BP2 microdeletion, as well as the genotypic spectrum of CSVD.
Acknowledgment
The authors would like to thank the RunningGene Inc. for technique assistant of whole exome sequencing and MLPA. Also, the authors thank Medsci for English language editing.
Footnotes
Disclosure. This work was supported by the National Natural Science Foundation of China, The Shanghai Natural Science Foundation Grant number (81971181)(19ZR1449200) and The Medical and Engineering Crossover Fund of SJTU Grant number (YG2017MS67) from Ailian Du.
- Received April 4, 2022.
- Accepted June 1, 2022.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.