


Abstract
Epilepsy, one of the most prevalent chronic neurological diseases, can cause severe morbidity as well as mortality. A mutation of the KCNMA1 gene results in a rare genetic disease that causes epilepsy as its core presentation. Both neurological and non-neurological manifestations have been reported in patients with KCNMA1 gene mutation. We are reporting a KCNMA1 gene variant referred to as c.2369C>T (p. Pro790Leu), which encodes the subunit of alpha of calcium-sensitive potassium channels, which causes epilepsy but not dyskinesia in a young Saudi female who is the daughter of consanguineous parents. Our case shows that calcium-sensitive potassium channels can cause an isolated generalized epilepsy as reported previously in a single case. Moreover, this case aids in delineating the clinical and structural picture and the treatment of the KCNMA1 gene mutation in patients.
In 2017, the International League Against Epilepsy (ILAE) accepted a change in terminology from the use of “idiopathic” to “genetic” when describing epilepsy syndromes.1 The KCNMA1 gene mutation is a rare epilepsy- associated gene with a non–well-defined prevalence. In this study, we report the rare case of a young female with KCNMA1 gene mutation to aid in the determination of the clinical features of this disease.
Case Report
Patient information
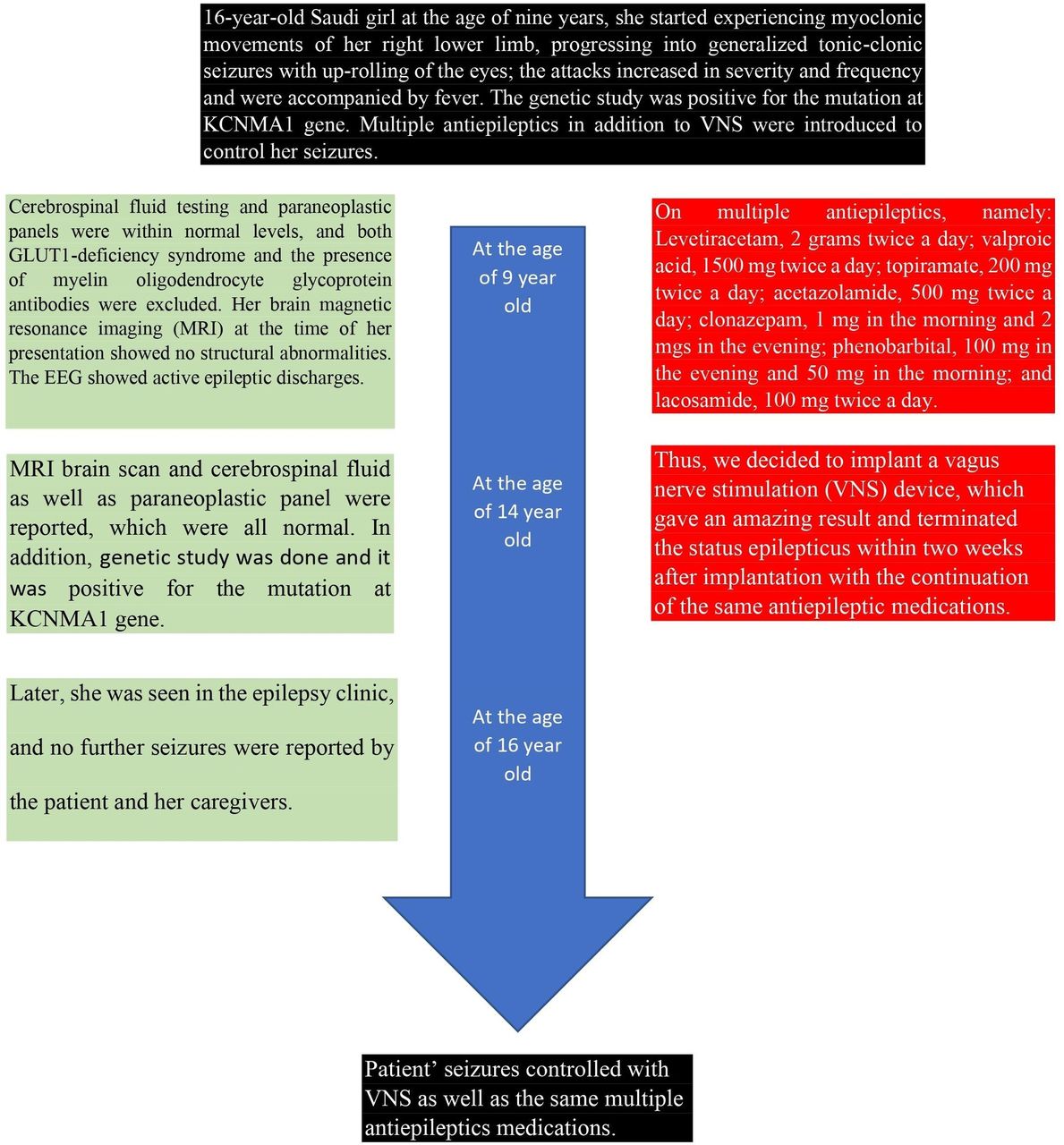
The 16-year-old, right-handed Saudi girl is the second child of consanguineous parents (Figure 1). Her mother’s pregnancy and delivery were unremarkable. Both her mother and older brother have generalized epilepsy. She had no history of any milestone delays, and her head circumference was within the normal percentile range from birth and throughout her growth. At the age of 9 years, she started experiencing myoclonic movements of her right lower limb, progressing into generalized tonic-clonic seizures with up-rolling of the eyes; the attacks increased in severity and frequency and were accompanied by fever. She was started on levetiracetam and topiramate, and her seizures were fairly well controlled for about 4 years. There was no history of limb weakness, slowness, or fatigability.

- Timeline showing the course of the presented patient during follow up and outcome.
Clinical findings
However, during follow-up with a pediatric neurologist at the age of 12 years, she was admitted to hospital with the suspicion of having Miller Fisher syndrome due to the presence of weakness, areflexia, and ophthalmoplegia, which improved after 5 doses of intravenous immunoglobulins.
Diagnostic assessment
Both cerebrospinal fluid testing and paraneoplastic panels were within normal levels, and both GLUT1-deficiency syndrome and the presence of myelin oligodendrocyte glycoprotein antibodies were excluded. Her brain magnetic resonance imaging (MRI) at the time of her presentation showed no structural abnormalities. The EEG which was done for her is shown in Figure 2. The results of the gene panel test are shown in Table 1.
- Results of exome sequencing gene panel test of a young Saudi female.

- The electroencephalogram illustrates electrographic seizure A) and B) started at the right Fronto-Polar2 (FP2) shortly associated with jerking movements of the left leg then become generalized.
Therapeutic intervention
Multiple anti-epileptic drugs (AEDs) were added later as the patient’s seizures were only partially controlled, and she was frequently admitted to the intensive care unit due to status epilepticus.
When the patient was seen in the adult neurology clinic at the age of 14 years, she was on the following AEDs: levetiracetam, 2 grams twice a day; valproic acid, 1500 mg twice a day; topiramate, 200 mg twice a day; acetazolamide, 500 mg twice a day; clonazepam, 1 mg in the morning and 2 mgs in the evening; phenobarbital, 100 mg in the evening and 50 mg in the morning; and lacosamide, 100 mg twice a day. She still suffered from frequent seizures for which an epilepsy monitoring unit was planned. Unfortunately, despite good compliance with the AEDs, a few months later, she presented to the ER with status epilepticus necessitating admission to the intensive care unit. Attempts to withdraw the anaesthetic drugs resulted in the re-emergence of status epilepticus; she stayed in the ICU for one month and was considered to be in super refractory status epilepticus. At an urgent meeting, the epileptologists, along with a paediatric neurologist who was following up with her, decided to repeat an MRI brain scan and cerebrospinal fluid and paraneoplastic panel, which were all normal. Thus, we decided to implant a vagus nerve stimulation (VNS) device, which gave an amazing result and terminated the status epilepticus within two weeks after implantation. She was extubated and kept on the same AEDs.
Follow-up and outcomes
Later, she was seen in the epilepsy clinic, and no further seizures were reported by the patient and her caregivers.
Discussion
The KCNMA1 gene mutation at 10q22.3, encoding the alpha subunit of calcium-sensitive potassium channels (BK channels)2 is a rare cause of generalized epilepsy and other neurological and non-neurological features. Both autosomal-dominant and autosomal-recessive mode of inheritance was a pattern, and neither functional variants studies nor studies of patient cells were conducted.2-6 The phenotype represented in our reported case, showing both isolated and frequent seizure events. The VNS implantation has been reported as an effective termination method for epilepsy-related channelopathies;7-8 however, the current case was the first to utilize VNS in the treatment of KCNMA1 mutation–related epilepsy, specifically. Furthermore, VNS can be used as a suppressor method for refractory status epilepticus related to this gene mutation. Further studies are needed to clarify the pathophysiology of this gene mutation disease and its common mode of inheritance so that an effective treatment can be found.
This study is limited by the fact that functional studies of the variants and studies of patient cells were not performed, and gene panel tests were not carried out for the patient’s family members. More experimental studies and clinical trials are required to verify these findings and to delineate the clinical and structural picture of KCNMA1 gene disease.
In conclusion, the KCNMA1 gene mutation is a rare cause of generalized epilepsy and other neurological and non-neurological features. The phenotype represented in our reported case, showing both isolated and frequent seizure events, supports previously published cases and influence the proposed controlling AEDS. Furthermore, VNS can be used as a suppressor method for refractory status epilepticus related to this gene mutation. Further studies are needed to clarify the pathophysiology of this gene mutation disease and its common mode of inheritance so that an effective treatment can be found.
Acknowledgement
The authors would like to thank the patient and her family for participating in this study. Also, we would like to thank Scribendi Inc (www.scribendi.com) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received April 8, 2022.
- Accepted August 9, 2022.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.