


Abstract
Dyke Davidoff- Masson syndrome is a rare disorder of hemiatrophy of the cerebral hemisphere and clinically manifests as hemiparesis, seizures disorder, mental retardation and facial asymmetry and has various perinatal or post natal etiologies and has characteristic radiological appearance on brain imaging. It is important to recognize the clinical and radiological features of this condition for the neurologists as well as the radiologists. We are discussing our case as this patient presented very late although his symptoms seem to present since his birth which is very unusual with this disorder.
Dyke-Davidoff-Masson syndrome (DDMS) is a rare disease comprising of hemiparesis, seizures, facial asymmetry, and mental retardation.1 The classical findings are of cerebral hemiatrophy and calvarial thickening along with hyperpneumatization of the frontal sinuses which are only present if an insult to the brain occurs before 3 years of age.2 Although, the symptoms are present in early childhood in most of the cases, but adult presentation has been reported by other authors as well.3,4 We are presenting this case, who had congenital hemiatrophy but his seizures started very late in the disease course and the diagnosis was confirmed by the CT head. As this case is unusual as patient with congenital hemiatrophy managed to have his activities of daily living, was not mentally retarded and seizures presentation was very late, hence, it highlights the importance of early suspicion and considering neuroimaging in such cases in the future.
Case Report
A 69-year-old male presented to our emergency department with a history of generalized tonic clonic seizures for the last 4-5 years, which have increased in frequency for the last 6 months. He was born out of a nonconsanguineous marriage. Birth history was indicative of a full-term normal delivery without any antenatal or perinatal complications but patient was noticed to have right sided hemiatrophy and hemiparesis at the time of birth but no medical record is available. According to patient siblings despite this right sided weakness, he had normal developmental milestones and was able to manage the activities of his daily living independently but was noticed to have hearing problem in early childhood but no medical attention was sought for any neurological problem so far.He is working as a plumber, has 4 brothers and one sister with no family history of any neurological illness.
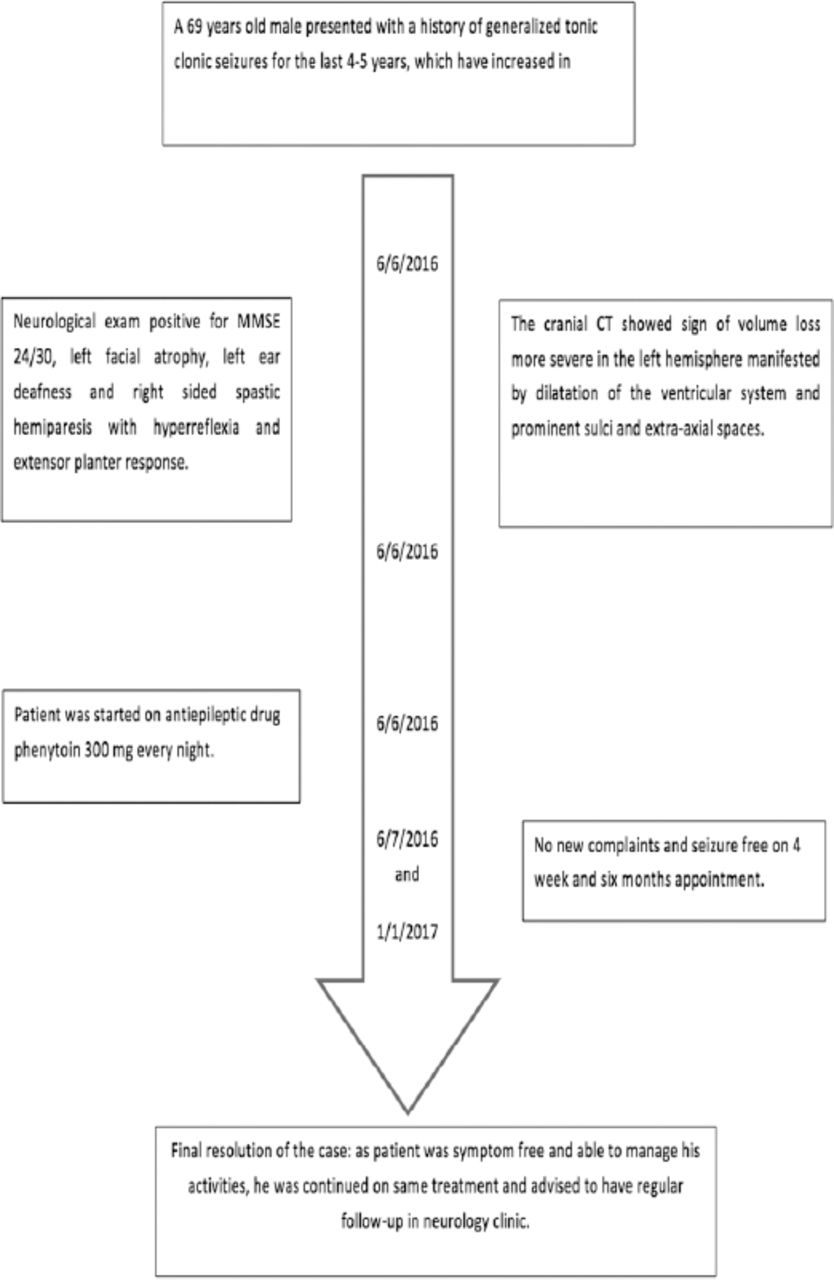
He started to have generalized tonic clonic seizures around 5 years ago, initially very infrequent, recurring every 6-8 months but became more frequent over the last 6 months with 4 episodes of generalised tonic clonic seizures during the last week that brought him to emergency department. There was no associated history of fever or mental status changes, so no suspicion of central nervous system infection arose. The timeline of patient illness and follow-up is presented in Figure 1.

The timeline of patient illness and follow-up is presented.
Clinical findings
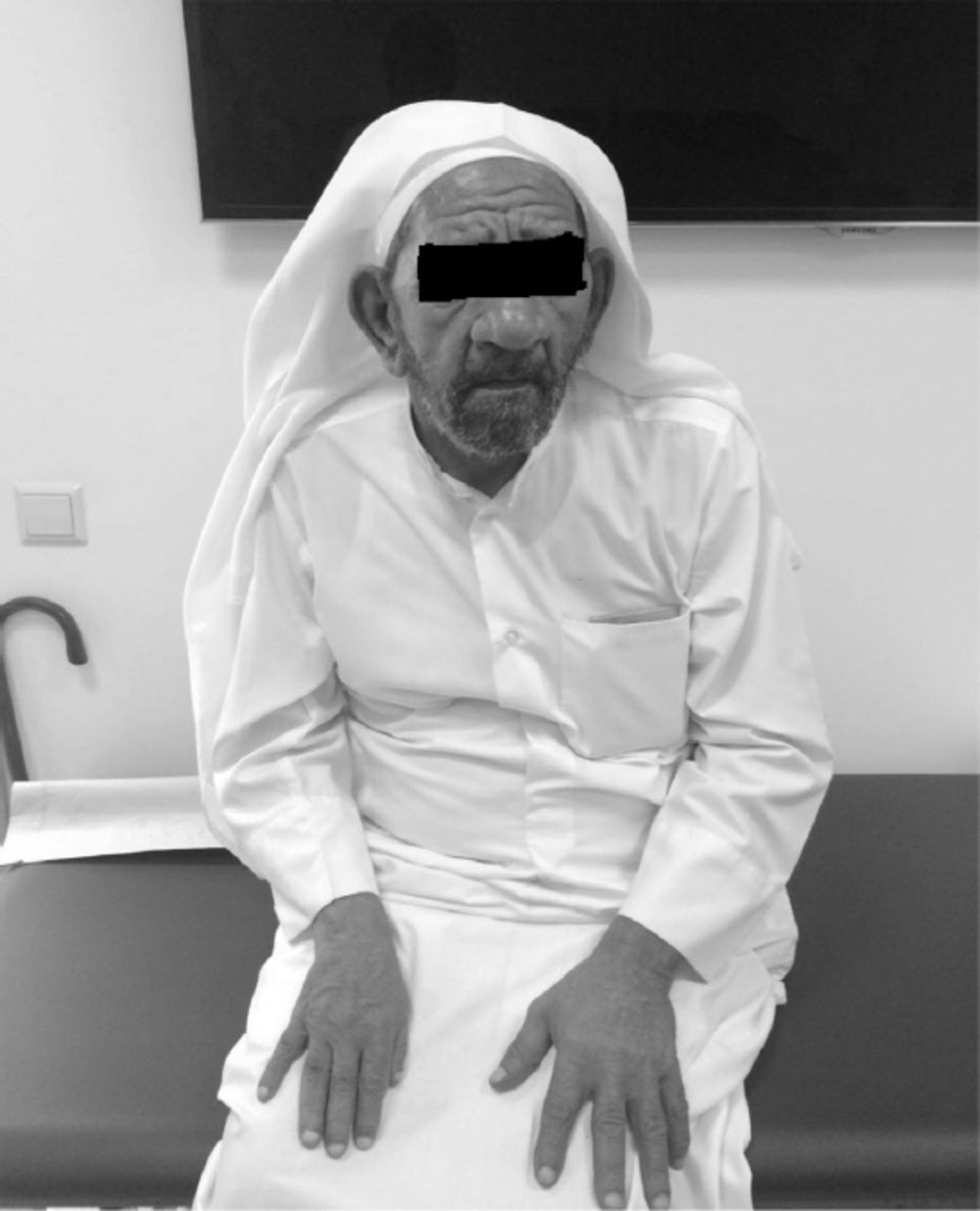
His examination did not reveal any manifestations of neurocutaneous disorder. His cognition and orientation was intact. He scored 24/30 on mini mental status examination, which was appropriate provided his poor educational background. There was mild left facial atrophy, left ear deafness and could barely hear from his right side but rest of the cranial nerve exam was unremarkable. His motor system exam revealed right sided spastic hemiparesis of grade 4/5 medical research council with hyperreflexia and extensor planter response. His gait was also hemiplegic (Figure 2).

Picture of the patient showing right sided facial and body atrophy.
Diagnostic assessment
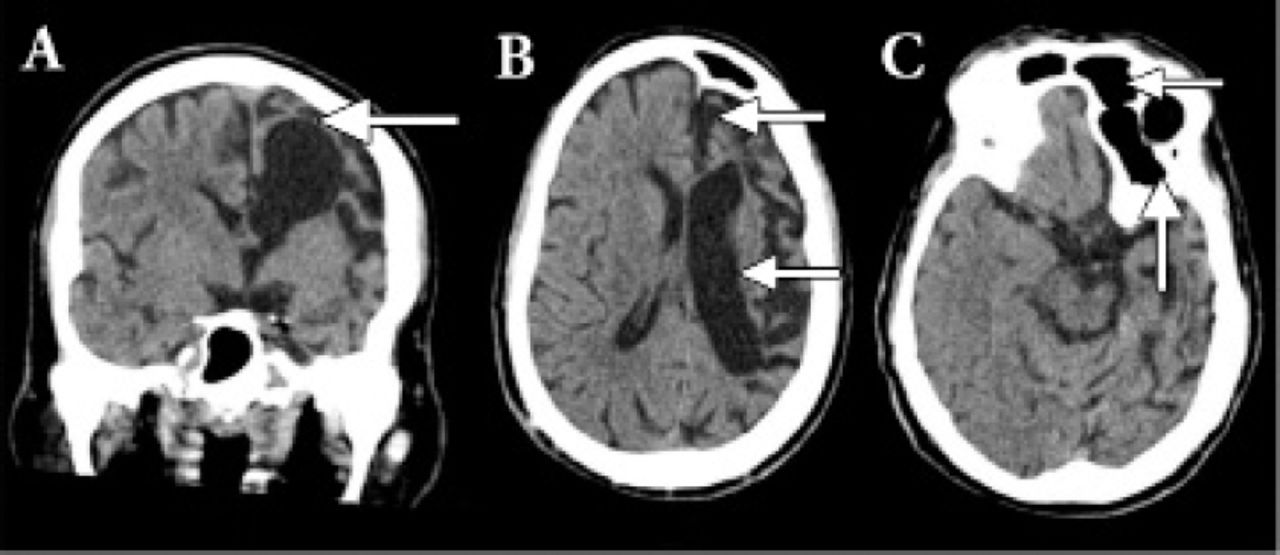
All the baseline metabolic and septic workup turned out to be negative as the cause of his symptoms. Patient underwent computerized topography from the Emergency department. The cranial CT showed sign of volume loss more severe in the left hemisphere manifested by dilatation of the ventricular system and prominent sulci and extra-axial spaces, consistent with left cerebral hemiatrophy. There is also enlargement of the left frontal air sinus with falcine displacement to the right side (Figure 3). Correlation with the patient’s clinical history of congenital anomaly since birth and facial asymmetry, this might represent Dyke-Davidoff-Masson syndrome. MRI brain was planned but could not be performed as patient had history of some dental braces that was not compatible with the MRI.
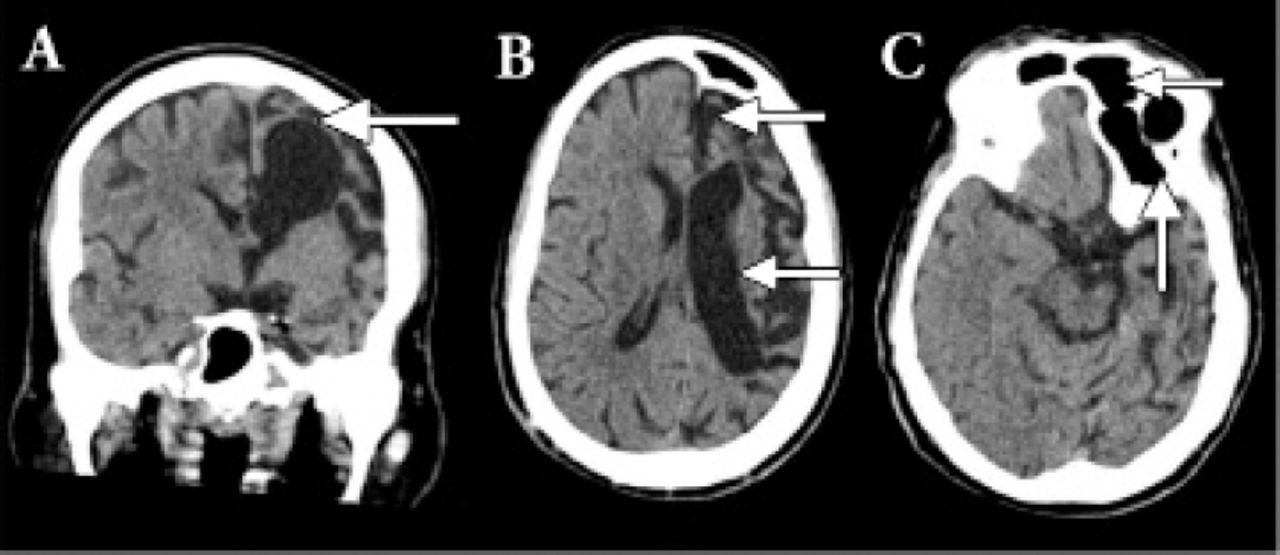
CT head of the brain showed sign of volume loss more severe in the left hemisphere manifested by dilatation of the ventricular system and prominent sulci and extra-axial spaces, consistent with left cerebral hemiatrophy.
Therapeutic intervention
Patient had generalized seizures and was diagnosed with epilepsy, he was started on single antiepileptic tab phenytoin 300 mg at night.
Follow-up and outcome
No new clinical findings were observed during follow-up examinations, patient remained seizure free when seen in the follow-up clinic after 4 weeks and then 6 months. Patient was encouraged to continue treatment.
Discussion
This is a rare condition first described by Dyke, Davidoff, and Masson in 1933 in a report of plain skull radiographic changes in 9 patients who presented with seizures, hemiparesis, facial asymmetry, and mental retardation.1 The clinical features include contralateral hemiparesis along with an upper motor neuron facial weakness, focal or generalized seizures, and mental retardation with learning disabilities.5 Usually, there is no sex predilection, and any side of the brain can be involved, although involvement of the left side and male gender have been shown to be more common in one study.2 Cerebral hemispheric growth is asymmetrical and there is atrophy or hypoplasia of one side with midline shift, ipsilateral osseous hypertrophy with hyperpneumatization of sinuses mainly frontal and mastoid air cells with contralateral paresis.6 Other features are enlargement of ipsilateral sulci, dilatation of ipsilateral ventricle and cisternal space, decrease in size of ipsilateral cranial fossa, and unilateral thickening of skull, these Changes can occur only when brain damage is sustained before 3 years of age, but it may become evident as early as 9 months after the injury.7 It can present in 2 forms, infantile (congenital) or acquired.8 The infantile variety usually results from the perinatal infections, coarctation of the midaortic arch or neonatal or gestational vascular occlusion involving the middle cerebral artery and symptom start in early perinatal or infantile period.8 While the acquired causes are trauma, infection, ischemia, tumor, hemorrhage or prolonged seizure. Age of presentation depends on the time of insult characteristic changes may only be seen in adolescent and adults.
In our patient, the volume loss is severe in the left side manifested by dilatation of the ventricular system and prominent sulci and extra-axial spaces and presence of hemiatrophy at the time of birth as reported by the patient’s relative reflected an onset of brain insult after the completion of sulci formation, probably of vascular origin involving left middle cerebral artery but there is no history of any antenatal or perinatal complications. Patients with congenital hemiatrophy present with early onset seizures which are refractory to treatment,9 but our patient presented at a very late age and his seizures were controlled with single antiepileptic medication.
The differential diagnoses include chronic Rasmussen encephalitis which is the chronic, progressive inflammation of brain of uncertain etiology and sturge weber syndrome. However, there are no calvarial changes in Rasmussen encephalitis and enhancing pial angiomas and cortical calcifications are the additional features noted in Sturge-Weber syndrome.9
Management consists of control of seizures with appropriate antiepileptic medications, as most patients with this disorder present with refractory seizures. Additionally, regular physiotherapy, occupational and speech therapy have a vital role in the management of the symptoms. Hemispherectomy is indicated in patients who have weakness or hemiplegia and refractory seizures and is successful in 85% of the cases. Prognosis is usually poor in cases of prolonged or recurrent seizures and if hemiparesis occurs before two years of age. Therefore, it is indeed important for neurologists, pediatricians and radiologists to be familiar with this condition for its early diagnosis and treatment.9 In conclusion although patients with DDMS present in early childhood or adolescents, but patient might present later as well, so early recognition is important as late diagnosis is associated with poor prognosis.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received May 11, 2017.
- Accepted March 14, 2018.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.