


Abstract
Kearns-Sayre Syndrome (KSS) is a subtype of chronic progressive external ophthalmoplegia (CPEO). In this case, A 21-year-old man diagnosed with KSS, and presented with chronic progressive blepharoptosis (ptosis) and external ophthalmoplegia, diffuse depigmentation of the retinal pigment epithelium, and cerebellar ataxia, with a cerebrospinal fluid protein of 254 mg/dL, was reported. Genetic screening revealed a novel mutated gene in SLC25A4 in the patient as well as in his mother: NM_001151:c.170G>C in exon 2. Its imaging finding is a characteristic progressive atrophy of the right cerebellar hemisphere. In conclusion, we found a case of KSS with a novel mutated gene in SLC25A4: NM_001151:c.170G>C in exon 2 as the pathogenic mechanism, and found that KSS can be caused only when the proportion of mutations in the SLC25A4 gene reach a certain degree, and the patient with KSS showed a unique cranial imaging feature of unilateral progressive cerebellar atrophy.
Kearns-Sayre Syndrome (KSS) is a subtype of chronic progressive external ophthalmoplegia (CPEO). The 3 main symptoms of KSS are onset before 20 years of age, CPEO, and pigmented retinopathy. At least one of the following symptoms is accompanied: complete heart block, cerebrospinal fluid protein greater than 100 mg/dL, cerebellar ataxia, short stature, deafness, dementia, and endocrine abnormality.1 Since Kearns and Sayre first reported KSS in 1958, there have been reports of mtDNA deletions at different locations and sizes in many patients.2 The most common deletion is marked as the “common 4977 bp deletion,” accounting for more than one-third of all cases.3-5 The central nervous system (CNS) involvement of the disease is reflected in the degree of MRI abnormalities, including hyperintensity of fluid-attenuated inversion recovery (FLAIR) sequence in the brainstem, globus pallidus, thalamus, and cerebral and cerebellar white matter. Here, we report a case of a 21-year-old man with KSS who had a novel mutated gene in SLC25A4: NM_001151:c.170G>C in exon 2 and had a special imaging finding of progressive atrophy of unilateral cerebellum.
Case Report
Patient information
The patient is 21-year-old male.
Diagnostic assessment
At the age of 13 years, parents of the patient brought him to a local children’s hospital due to a speech rate lower than that of a normal child, with no abnormality found on the children’s intelligence screening test. Cranial MRI revealed (A1 and C1 in Figure 1) obvious cerebellar vermian sulcus, and mild atrophy was not excluded. His symptom slowly worsened, and significantly worsened at the age of 16 years, specifically with significantly elongated speech, heterogeneous pitch, and often fulminant. He visited a local hospital and cranial MRI revealed (A2, B1 and C2 in Figure 1) revealed slight narrowing of the gyri in the right cerebellar hemisphere and mild atrophy of the right cerebellum, and the above symptoms gradually worsened. At the age of 19, the patient gradually developed right blepharoptosis, inability to move in the lower and right lower direction (Figure 3), and double vision, with significant elongated speech, heterogeneous pitch, and often fulminant. He visited our Neurology Department at the age of 21 (Table 1).
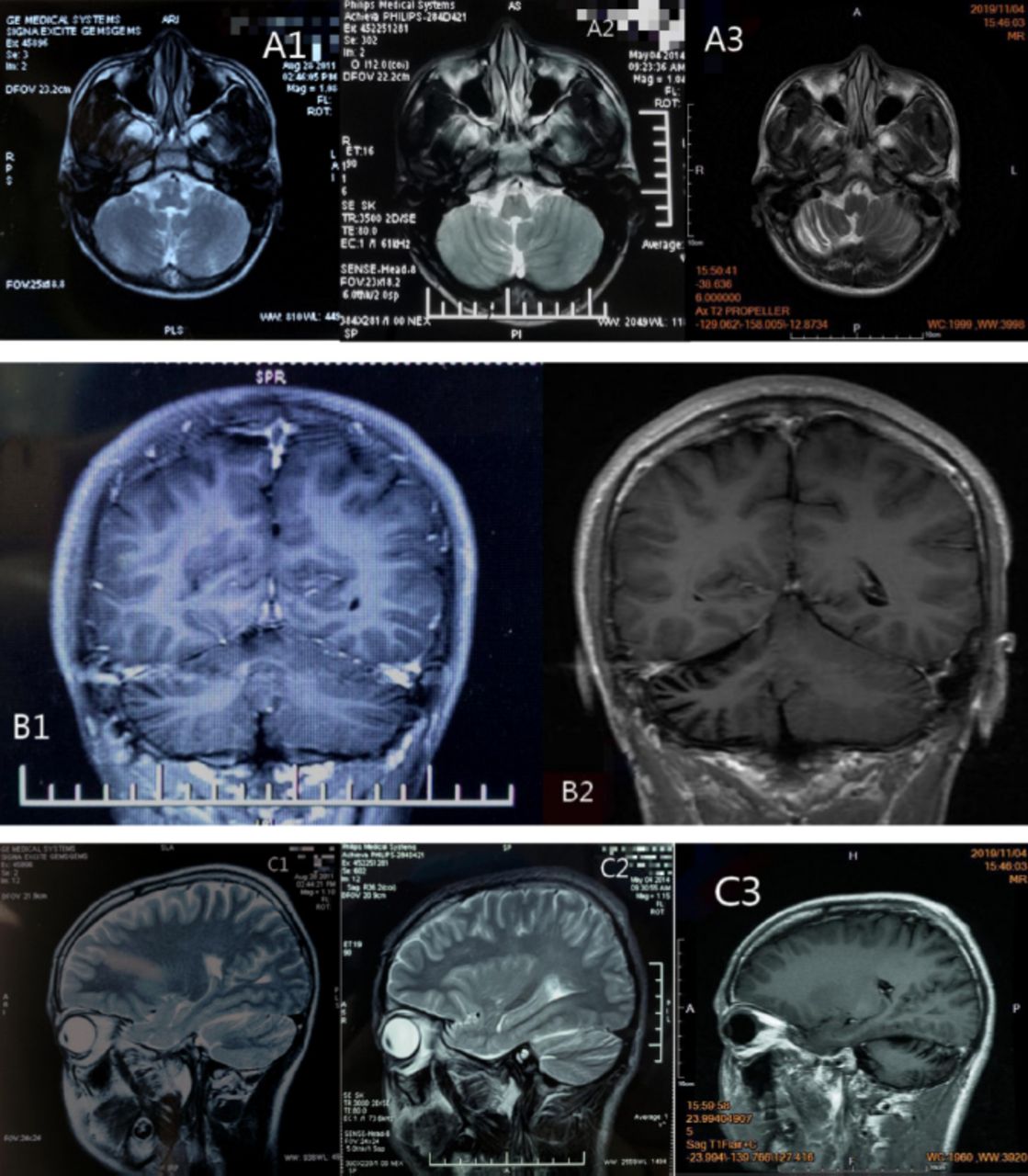
- MRI images A1 and C1) are the cranial MRI images on May 14, 2011. They reveal obvious sulcus of vermis cerebelli; A2, B1 and C2) are the cranial MRI images on August 4, 2014. They reveal slight narrowing of the gyrus of the right cerebellar hemisphere; A3, B2 and C3) are the cranial MRI images on November 4, 2019. They reveal narrowing of the gyrus of the right cerebellar hemisphere and widening of the sulcus.

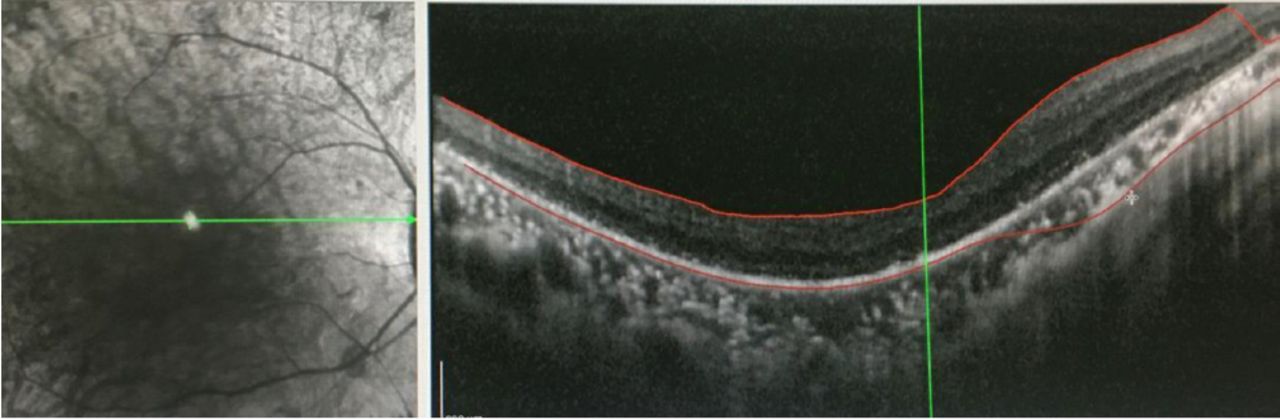
- Outer retinal atrophy in the right eye revealed by OCT of both eyes in the Department of Ophthalmology of our hospital in November 2019.

- The physical examination image A) November 1, 2019: both pupils are of the same size and roundness, with a diameter of about 3mm, sensitive to light reflex, blepharoptosis for the right, and mild restriction for movement in the downward and lower right directions; B) April 20, 2020: both pupils are of the same size and roundness, with a diameter of about 3 mm, sensitive to light reflex, blepharoptosis for the right, unable to move in the downward and lower right directions.
- Patient timeline.
Clinical findings
Results of laboratory test, electrophysiological examination, cerebrospinal fluid examination, ECG and other related examinations are provided in the table (Table 2). The OCT showed outer retinal atrophy in the right eye (Figure 2), cranial MRI showed significant atrophy of the right cerebellar hemisphere (A3, B2 and C3 in Figure 1), and cranial contrast-enhanced MRI did not show significant abnormalities. The test result of mitochondrial gene hotspot mutation in MELAS (mitochondrial encephalomyopathy) syndrome using PCR-Sanger was negative (Table 3). The result of copy number variation sequencing (CNV-Seq) using PCR-Sangers sequencing was negative, the test of mitochondrial disease-related nuclear genome using next-generation sequencing (NGS, whole exome sequencing analysis) showed that the patient carried the suspected pathogenic gene of SLC25A4. Then the first-generation sequencing for SLC25A4 gene was performed on his parents, and the results indicated that the NM_001151:c.170G>C heterozygous mutation of SLC25A4 gene occurred in the exon 2 region in both the patient and his mother. Quantitative polymerase chain reaction5 revealed that the patient had mitochondrial DNA deletion (mitochondrial/nuclear DNA: 18.7%) (mitochondria DNA depletion (mito-chondria/nuclear DNA: 18.7%)), and the patient’s mother had mitochondria DNA depletion (mitochondria/nuclear DNA: 2.7%). This study was approved by the Ethics Committee of the Affiliated Hospital of Chengdu University of TCM.
- Laboratory examination, electrophysiological examination, cerebrospinal fluid (CSF) examination and other examinations.
- Negative test result of mitochondrial gene hotspot mutation in MELAS (Mitochondrial Encephalomyopathy) syndrome using PCR-Sanger.
Therapeutic intervention
The patient received trophic nerve therapy during hospitalization.
Follow-up and outcomes
The symptoms were not significantly improved. After discharge, the patient continued to be followed up for about half a year by the outpatient department. The above symptoms continued to worsen, without other clinical symptoms.
Discussion
The KSS is very rare. Although the exact prevalence of this disease is unknown, a study reported a prevalence of 1.6 cases per 100,000 people in the Finnish population.5 In this report, the patient had onset at the age of 13, with the clinical manifestations of chronic progressive blepharoptosis (ptosis) and external ophthalmoplegia, diffuse depigmentation of the retinal pigment epithelium, cerebellar ataxia and cerebrospinal fluid protein of 254 mg/dL, and the clinical features were consistent with the diagnosis of KSS.
Genetic screening revealed a novel mutated gene in SLC25A4 in the patient as well as in his mother: NM_001151:c.170G>C in exon 2. Several mutations in SLC25A4 are associated with mitochondrial diseases and are divided into 2 different clinical phenotypes: null recessive mutations leading to mitochondrial myopathy and cardiomyopathy phenotypes, appearing in childhood or early adulthood and characterized by fatigue and exercise intolerance (also known as mitochondrial DNA depletion) (type of syndrome 12 [MIM: 615418])6,7 and the condition of pathogenic autosomal dominant progressive external ophthalmoplegia caused by several separate heterozygous mutations in adult (adPEO [MIM: Report 609283])5,7 but the NM_001151:c.170G>C mutation in exon 2 in SLC25A4 has never been reported, and in combination with the clinical manifestations of the patient and his mother, we found that mutations at this site do not necessarily cause the development of KSS, and Sanger sequencing reveals NM_001151.4: c.239G > a (p.a arg80his) heterozygous de novo mutation in SLC25A4. Quantitative polymerase chain reaction5 revealed that the patient had mitochondrial DNA deletion (mitochondrial/nuclear DNA: 18.7%) (mitochondria DNA depletion (mito-chondria/nuclear DNA: 18.7%)), and the patient’s mother had mitochondria DNA depletion (mitochondria/nuclear DNA: 2.7%). We speculate that KSS can be caused only when the proportion of mutations in the SLC25A4 gene reach a certain degree.
Postmortem neuropathology in KSS patients may sometimes be associated with severe demyelination of white matter tracts in the brain.5,7 Central nervous system (CNS) involvement is reflected in the degree of MRI abnormalities, including hyperintensity of fluid-attenuated inversion recovery (FLAIR) sequence in the brainstem, globus pallidus, thalamus, and cerebral and cerebellar white matter.7 But these findings were not seen in this patient, but rather a significant progressive atrophy of the right cerebellar hemisphere, which has not been reported in previous cases of KSS.
- Received September 24, 2021.
- Accepted February 1, 2022.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}
{kind=link}
{kind=link}