


Abstract
Subependymal giant cell astrocytoma (SEGA) is a rare circumscribed astrocytic glioma that occurs in approximately 25% of all tuberous sclerosis (TSC) cases. Herein, we discuss an atypical presentation of SEGA, including the genetic alterations, impact on clinical presentation, and the determinants of each medical and surgical treatment option. A 14-year-old girl presented with intermittent headache and a right intraventricular mass originating near the foramen of Monro. The tumor’s proximity to critical structures necessitated maximum safe resection, which improved her symptoms. Histological findings indicated SEGA, and genetic sequencing revealed a TSC2 mutation. However, complete clinical and radiological evaluations failed to reveal TSC. Two months later, a new subependymal nodule was incidentally found. She had a recurrent left occipital horn lesion and diffuse smooth leptomeningeal enhancement with no spine drop metastases. She was administered everolimus as the tumor was considered unresectable. Subsequent imaging revealed a reduction in both residual and new lesions.
Subependymal giant cell astrocytoma (SEGA) is a rare central nervous system (CNS) tumor arising near the foramen of Monro that rarely extends to the third ventricle.1 It commonly manifests in the first 2 decades of life and rarely at the extremes of age.2,3,4 SEGA affects patients with tuberous sclerosis complex (TSC) and is most prevalent in those below 25 years of age.2 The TSC clinically manifests as multiple skin, eye, kidney, liver, and brain lesions.4 Mutations in the genes, TSC1 or TSC2, encoding hamartin and tuberin proteins, respectively, cause loss of inhibition over the mammalian target of rapamycin (mTOR), which normally control cell growth, proliferation, and metabolism.5 Herein, we report a case of SEGA lacking the clinical stigmata of TSC at presentation.
CASE Report
Patient information
This case report was approved by the institutional review board of XXX (XXX). Patient privacy and confidentiality were assured, no identifiers were collected, and all data are kept in a secure place within NGHA premises. Therefore, the requirement for informed consent was waived.
A 14-year-old girl with no previous medical history presented to the emergency department with a 6-month history of morning headaches, nausea, and vomiting.
Clinical findings
The patient was alert with stable vital signs and a Glasgow Coma Scale score of 15. Physical examination revealed loss of sensation over the distribution of the left maxillary nerve and bilateral papilledema. The latter resolved after shunting. Furthermore, visual acuity, fields, power, tone, and reflexes were all normal.
Diagnostic assessment
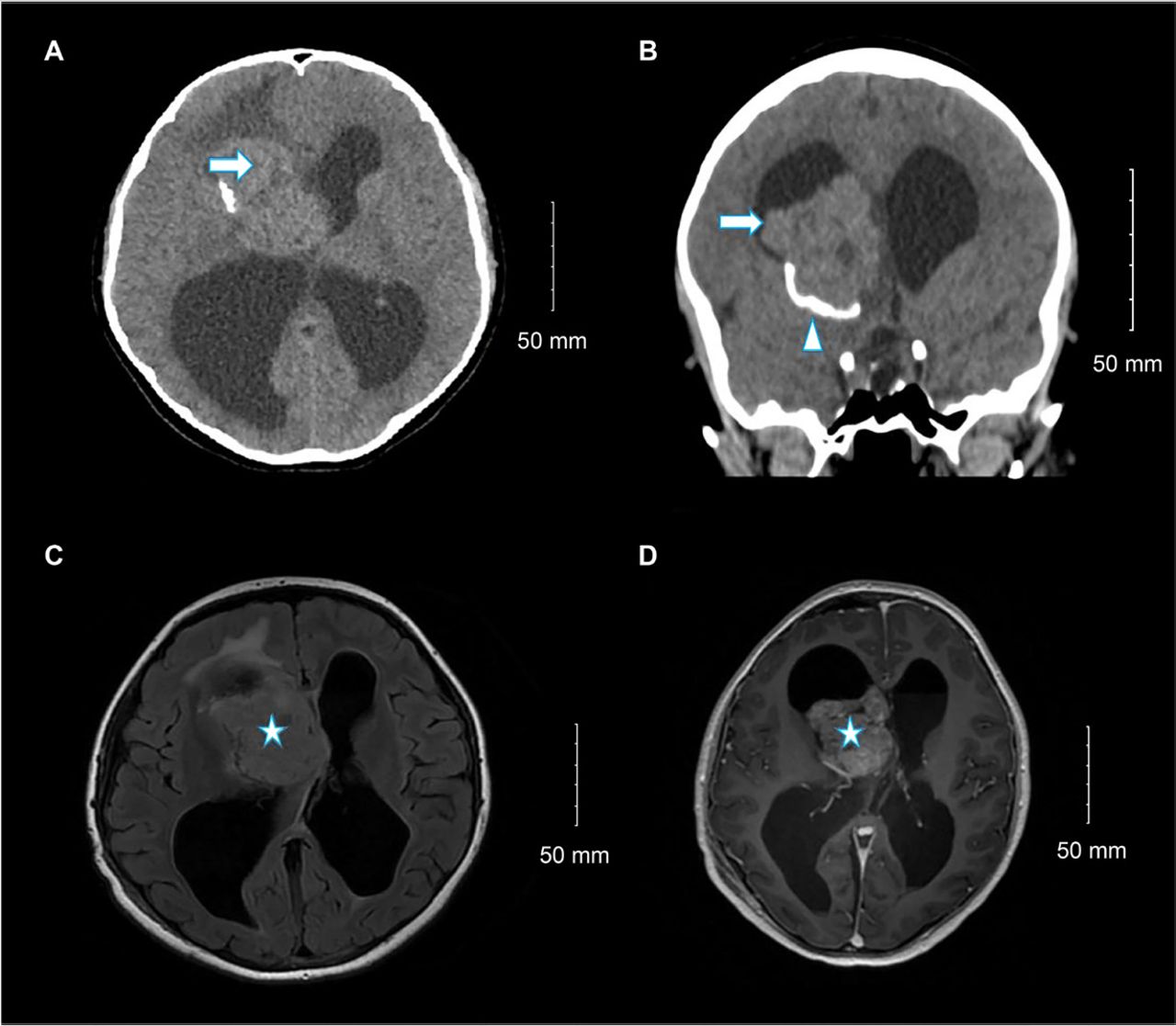
Non-contrast brain computed tomography (CT) showed a right lateral ventricular mass with calcification (Figure 1A-B). Subsequent contrast-enhanced magnetic resonance imaging (MRI) revealed a 6×5.3×3.3 cm well-defined intraventricular mass originating at the foramen of Monro and extending into the third ventricle. Hydrocephalus and left midline shift were observed (Figure 1C-D). The tumor appeared heterogeneous, with predominant iso-signal intensity on T1- and T2-weighted images and abnormal signal intensity in the cerebral cortex on fluid-attenuated inversion recovery imaging. Differential diagnoses included choroid plexus tumors and central neurocytoma.

- Selected (A) axial and (B) coronal images of unenhanced brain computed tomography show a large right lateral ventricle frontal horn dense mass (arrow) associated with peripheral calcification (arrowhead) causing marked hydrocephalus. Selected (C) axial fluid-attenuated inversion recovery and (D) axial T1 post-contrast brain magnetic resonance imaging show large, well-defined, lobulated intraventricular mass lesions within the right lateral ventricle at the region of foramen of Monro with heterogeneous moderate enhancement (star) and hydrocephalus.
Therapeutic intervention
An external ventricular drain (EVD) was inserted to alleviate hydrocephalus and the patient was transferred to the pediatric intensive care unit (PICU). A right frontal craniotomy with maximum safe resection was performed. An intraoperative diagnosis of subependymal giant cell astrocytoma was made. The EVD was kept in place postoperatively.
Pathological findings
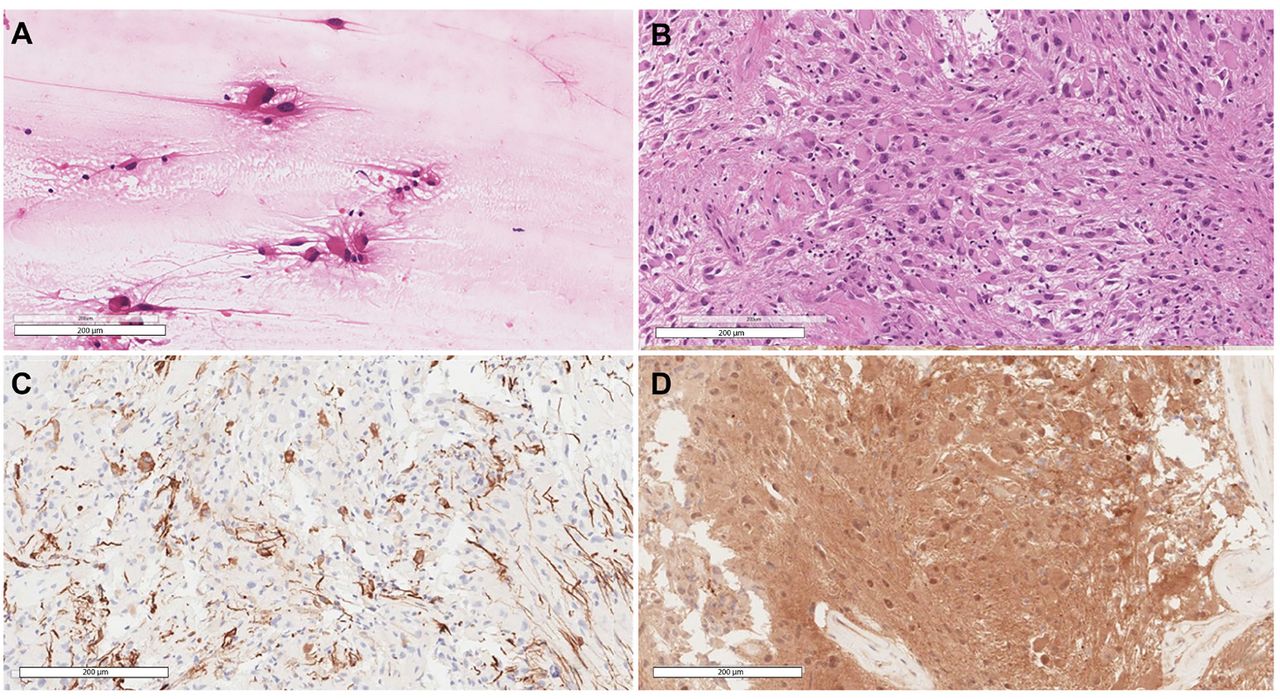
The histomorphology was typical of SEGA, depicting the polygonal glial cells with glassy eosinophilic cytoplasm, plump nuclei, and occasional ganglion-like cells (Figure 2A-B). There were no high-grade features, therefore, a CNS WHO grade 1 was assigned. The tumor cells were immunoreactive for glial fibrillary acidic protein, S100, and thyroid transcription factor-1, while negative for Olig-2, epithelial membrane antigen, and NeuN. Tumor cells’ DNA was extracted and followed by Extended Neuro-Oncology Gene Panel sequencing at Mayo Clinic Laboratories which revealed a pathogenic TSC2 mutation, c.119G>T:p.Q373H (Pro373His). Whole-genome sequencing of the patient’s blood sample was negative, indicating a sporadic tumor.

- Selected views of A) Smear slide at 20× showing the large eosinophilic tumor cells with processes emanating from them. B) Hematoxylin and eosin-stained section at 20× power reveals the classic histomorphology of subependymal giant cell astrocytoma with some of the cells showing well-defined borders while others show elongated cytoplasmic processes. C) Glial fibrillary acidic protein stain highlighting scattered positive tumor cells. D) Positive immunohistochemical staining for S100.
Follow-up and outcomes
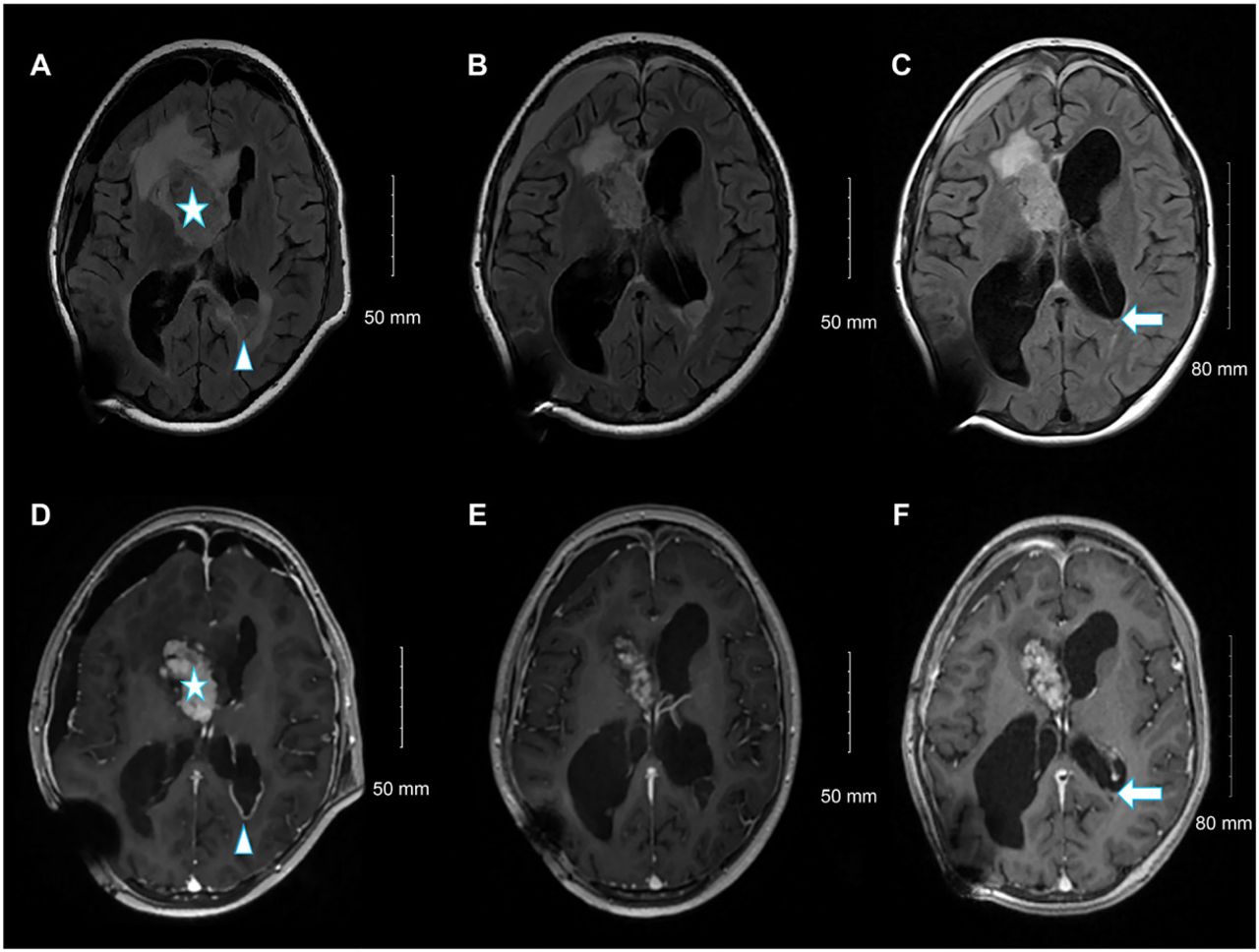
Postoperatively, the patient experienced multiple episodes of tonic-clonic seizures and was administered levetiracetam. Her symptoms improved dramatically, and she was discharged on postoperative day 21. Clinical/radiological surveys revealed no TSC stigmata. The EVD was then replaced with a ventriculoperitoneal shunt. Two months later, the patient developed a subdural hematoma and a new left occipital subependymal nodule measuring 2.7 cm, raising the possibility of transependymal cerebrospinal fluid (CSF) spread (Figure 3A-B). Therefore, everolimus, an mTOR inhibitor, was administered at a starting dose of 4.5 mg/m2 per day, which was increased to 5 mg for 2 weeks, followed by a further increase to 7.5 mg. Follow-up MRI at 3 and 6 months showed a reduction in the size of the residual tumor and occipital nodule (Figure 3C-F); no adverse effects were observed (Table 1).

- From left to right, selected fluid-attenuated inversion recovery magnetic resonance images (A, C, and E), and selected axial T1 post-contrast images at the same level (B, D, and F). Upon admission (A, B), the residual heterogeneously enhanced the right lateral ventricle frontal horn component (star). A new left occipital ependymal/subependymal nodule with peripheral enhancement (arrowhead) is visible. Follow-up examination at 3 months (C, D) revealed an interval reduction in the size of the left occipital ependymal/subependymal nodule, with peripheral enhancement. Follow-up examinations at 6 months (E, F) showed a further reduction in the size of the left occipital ependymal/subependymal nodule (arrow).
- Timeline table.
Discussion
Although SEGA is rare, it should be considered when encountering masses near the foramen of Monro in the pediatric population.6 Most patients with SEGA harbor an asymptomatic tumor. Consequently, the surveillance imaging protocols and recommendations from the International TSC Consensus Guidelines enable their detection.4 In other instances, SEGAs can present acutely with life-threatening obstructive hydrocephalus or intratumoral hemorrhage. The SEGAs have been regarded as almost exclusively pathognomonic of TSC; therefore, the stigmata of TSC must be searched in patients diagnosed with SEGA at presentation.2
Cases with no cognitive disability or those who lack tuberous manifestations at initial diagnosis are called forme fruste. The TSC forme fruste has been proposed for solitary SEGAs, manifested by somatic mosaicism rather than germline mutations.3 Additionally, Ichikawa et al7 reported a case of SEGA with two somatic hits of TSC2.
Parental testing for signs of TSC is integral for suspecting sporadic cases. Further genetic testing for somatic mosaicism is warranted to confirm whether this is a de novo event. In a series of 62 unrelated families with mutated TSC genes, one family had three children with TSC mutations. Further testing of seemingly asymptomatic parents revealed gonadal mosaicism.4 Detecting low levels of mosaicism can be challenging even with the best genetic testing methods. Therefore, screening the patient’s family for the mildest TSC manifestations is crucial for genetic counseling.8
In this case there were no TSC stigmata, however, the patient developed a new left occipital horn nodule and leptomeningeal enhancement along the spine, raising concerns regarding CSF spread. However, previous spinal imaging and CSF cytology samples were unremarkable. This lesion was then described as a subependymal nodule (SEN), a common manifestation of TSC, along with cortical tubers. Given the short interval since the emergence of this new nodule and the fact that TSC2 variants are associated with more severe manifestations, follow-up for metastasis or disease progression was considered.6,5
Surgery is the standard option for symptomatic SEGA, but some factors may favor alternative treatment.6 In this case, the tumor’s proximity to critical structures like the fornix and caudothalamic area favored the use of an mTOR inhibitor (everolimus). Clinical evidence suggests that everolimus is an effective treatment, with a significant reduction in tumor volume and a reduction in SEGA-related seizure frequency.9-10 The optimal dose is 4.5 mg/m2. Given the tumor’s benign nature, we decided to continue medical therapy until it is ineffective. Everolimus achieved significant reductions in the SEN and SEGA remnants.
Acknowledgments
We would like to thank Fahd M. Al Sufiani, Reema A. Alwabel, and Ghada A. Aldihan for their support. We would like to thank Editage (www.editage.com) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.