


Abstract
Spinocerebellar ataxia type 8 is a progressive neurodegenerative disease induced by expansion of CTA/CTG repeats in an untranslated region of the ATXN8/ATXN8OS gene. We report an elderly female patient presenting with rigidity, bradykinesia, ataxia and oculomotor defect at the disease onset age of 65 years old without family history, and hummingbird sign in cranial MRI, initially diagnosed as progressive supranuclear palsy (PSP). But genetic test showed that one allele of ATXN8OS gene had more than 131 CTA/CTG repeats which was a full penetrance mutant. It’s possible that this is a case of PSP with an ATXN8OS gene mutation that doesn’t contribute to the phenotype. Whether the ATXN8OS gene CTA/CTG repeats cause PSP phenotype needs further investigation with larger samples and pathological findings.
Spinocerebellar ataxia type 8 (SCA8) is a progressive neurodegenerative disorder resulting from the expansion of CTA/CTG repeats in an untranslated region (UTR) of the ATXN8/ATXN8OS gene. It is clinically characterized by a gradual onset and slow progression, presenting with varying degrees of cerebellar ataxia that may be accompanied by oculomotor disturbances, pyramidal signs, extrapyramidal signs, dysarthria, dysphagia, cognitive impairment and neuropsychiatric symptoms.1 Progressive supranuclear palsy (PSP) is a complex clinicopathologic condition that affects individuals in their mid-60s, characterized by postural instability, vertical supranuclear gaze palsy and frontal lobe dysfunction, and can be definitely diagnosed through the presence of 4 repeat tau neuropathology.2 Clinically, the occurrence of PSP in patients with SCA is exceedingly rare. This study provides further evidence supporting the PSP phenotype associated with abnormal CTA/CTG repeats in ATXN8OS gene.
Case Report
Patient information
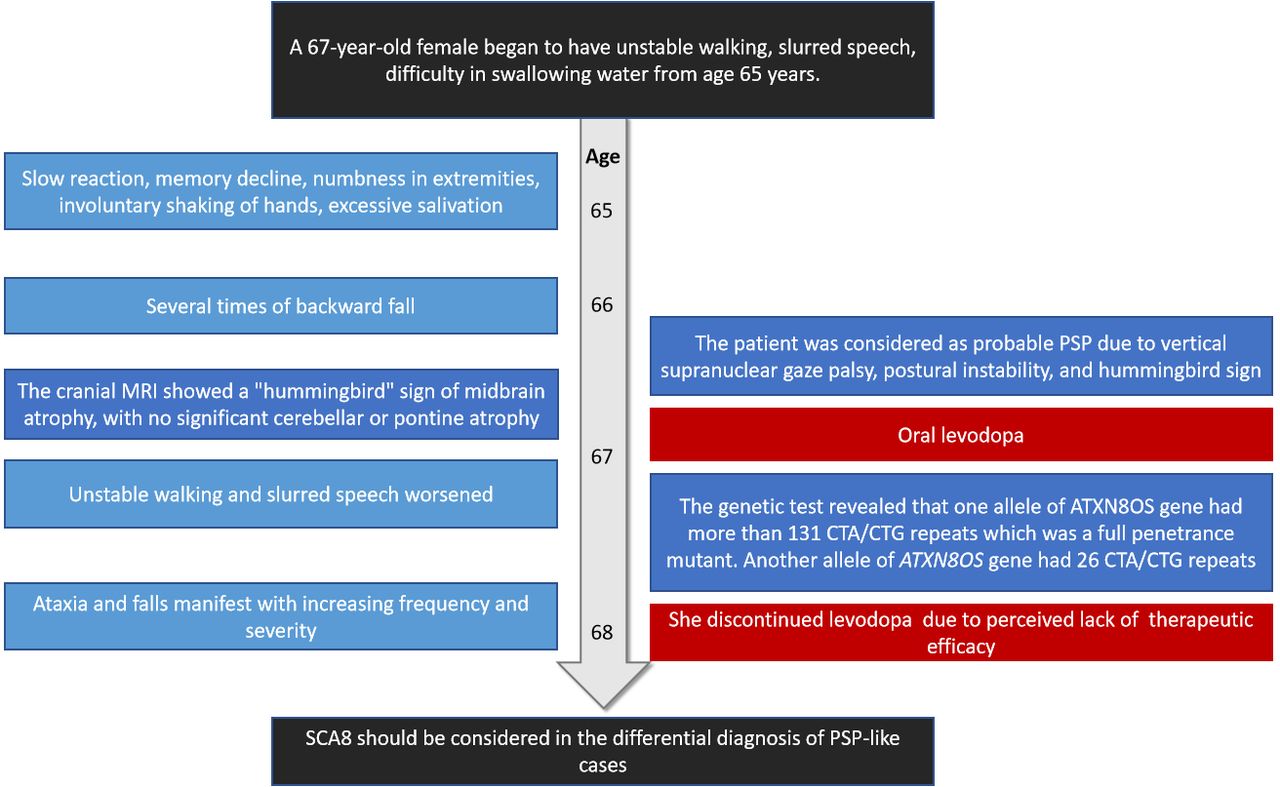
A 67-year-old female began to have unstable walking, slurred speech, difficulty in swallowing water from age 65 years. Slow reaction, memory decline, numbness in extremities, involuntary shaking of hands, excessive salivation were also reported. She denied night terrors, impaired urination and defecation. Her symptoms gradually worsened, and she had several times of backward fall. She had a history of hypertension and was taking anti-hypertensive medication regularly. She denied other long-term medication history and toxic substance exposure. She was educated for 6 years. Her spouse and 2 daughters were in good health. Her parents did not have symptoms of unstable walking before they died. Consanguineous marriage was denied. She second elder brother reported history of brain tumor, and other siblings were healthy (Figure 1A).
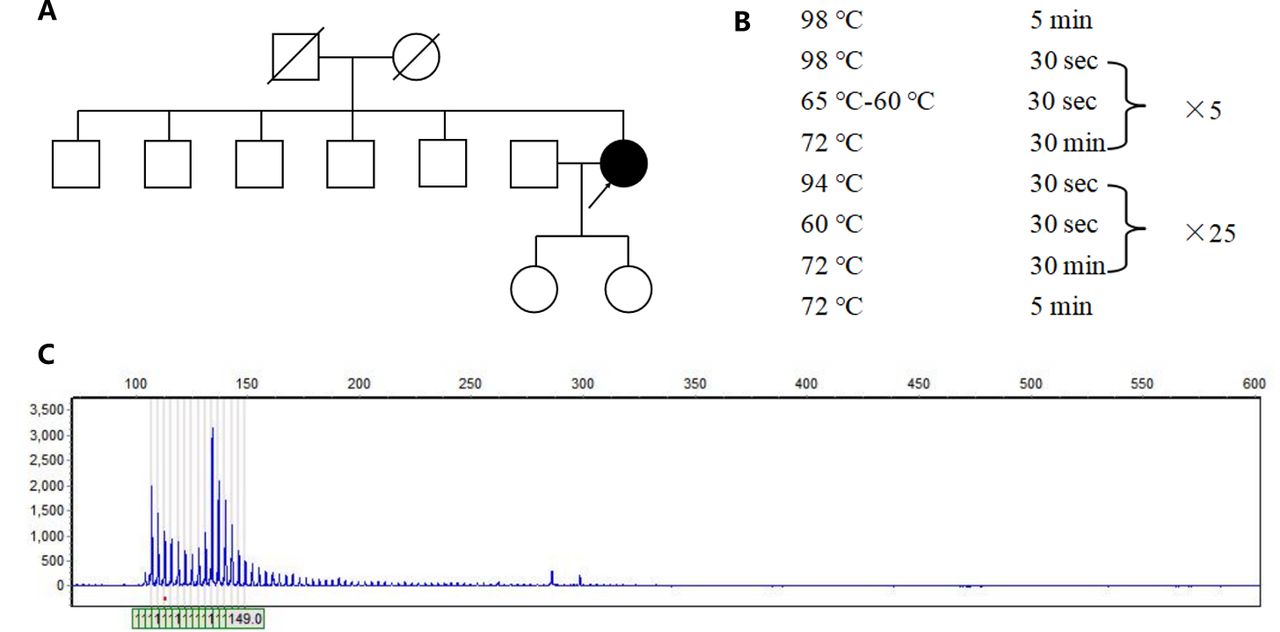
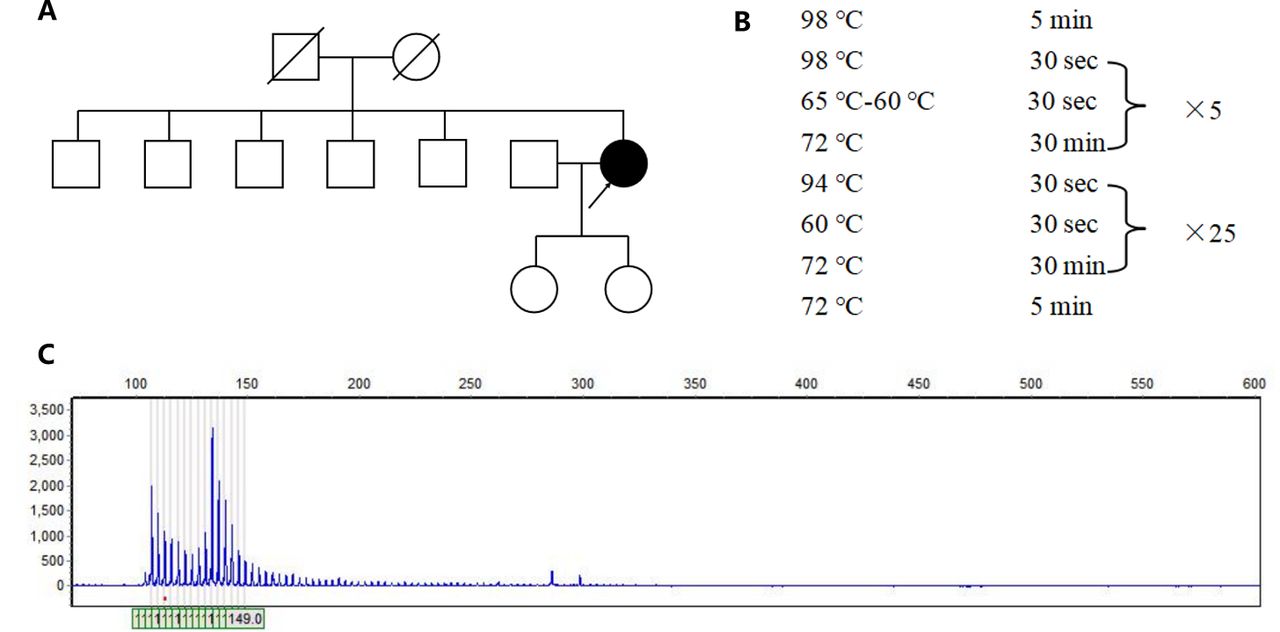
- Genetic analysis of the patient. A) The pedigree of the patient’s family. B) The amplification procedure of polymerase chain reaction. C) The genetic test revealed that one allele of ATXN8OS gene had more than 131 CTA/CTG repeats.
Clinical findings
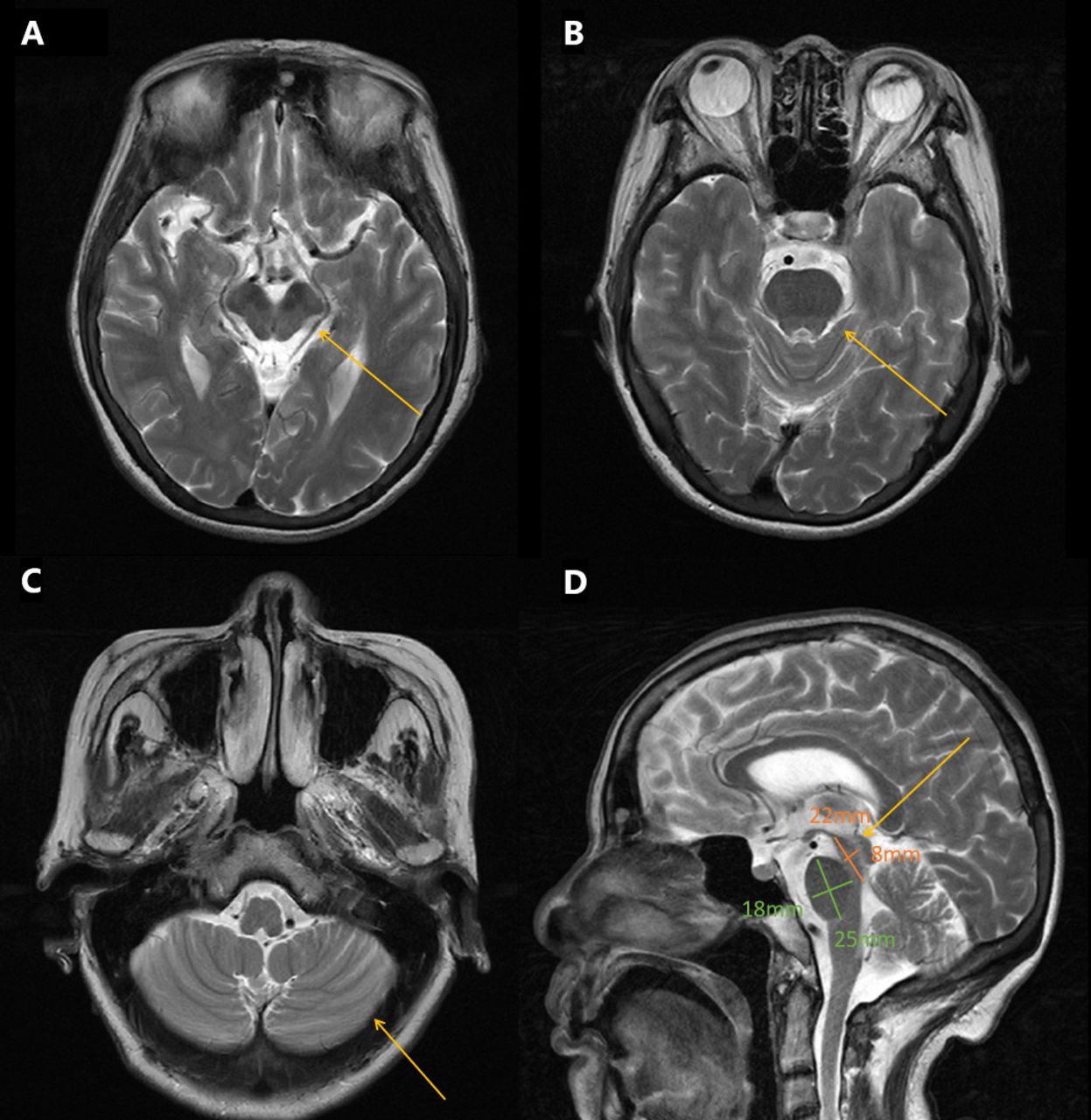
Neurological examination revealed mild dysarthria, bradykinesia and ataxic gait. She had difficulty opening her eyes and limited vertical movement of bilateral eyeballs. We observed rigidity mainly on the right side, slight bilateral upper limb postural tremor, and hyperactive tendon reflexes in the extremities (++++). Bilateral Babinski’s sign and Romberg’s sign were negative. Levodopa challenge test showed that the improvement rate of MDS-UPDRS (the Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale) part III was 15.8% (baseline score 38) after oral administration of levodopa 200 mg. Neuropsychological evaluations revealed a degree of cognitive impairment, with scores of 19 out of 30 on the mini-mental state examination and 8 out of 30 on the Montreal Cognitive Assessment. The cranial magnetic resonance imaging (MRI) showed a “hummingbird” sign of midbrain atrophy, with no significant cerebellar or pontine atrophy (Figure 2). The midbrain to pons ratio was 0.44 (<0.52) (Figure 2D), suggesting PSP with high sensitivity and specificity.2 No significant abnormal signals were seen in susceptibility weighted imaging (not shown). No spinal cord atrophy was seen on thoracic spine MRI (not shown). The progression of patient symptoms, diagnosis, and treatment was showed in Figure 3.
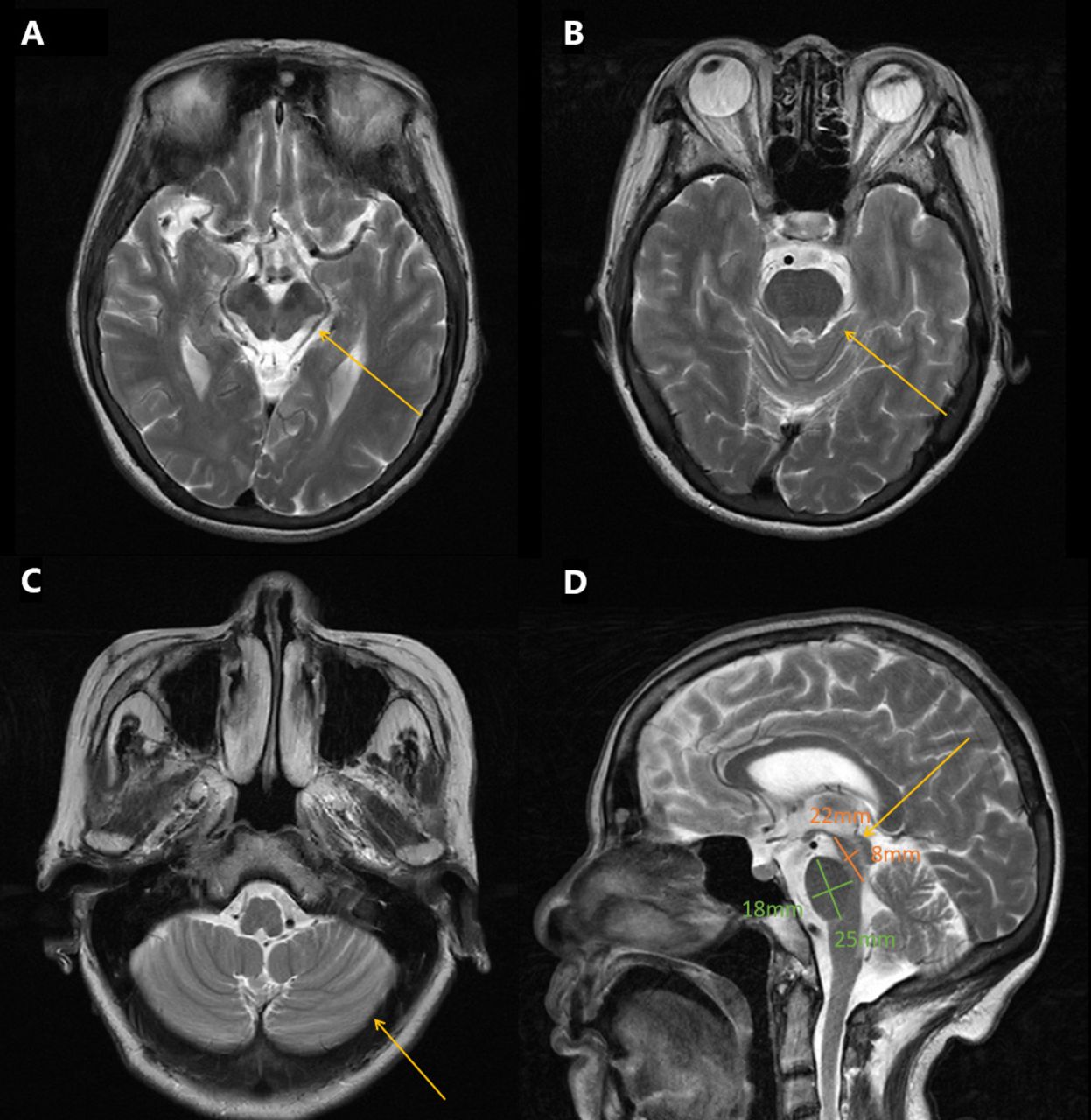
- Brain MRI T2-weighted images of the patient. A) Axial brain MRI demonstrated midbrain atrophy B) No significant pontine atrophy. C) No significant cerebellar atrophy. D) Saggital image revealed hummingbird sign, the midbrain to pons ratio was 0.44 (yellow arrows).
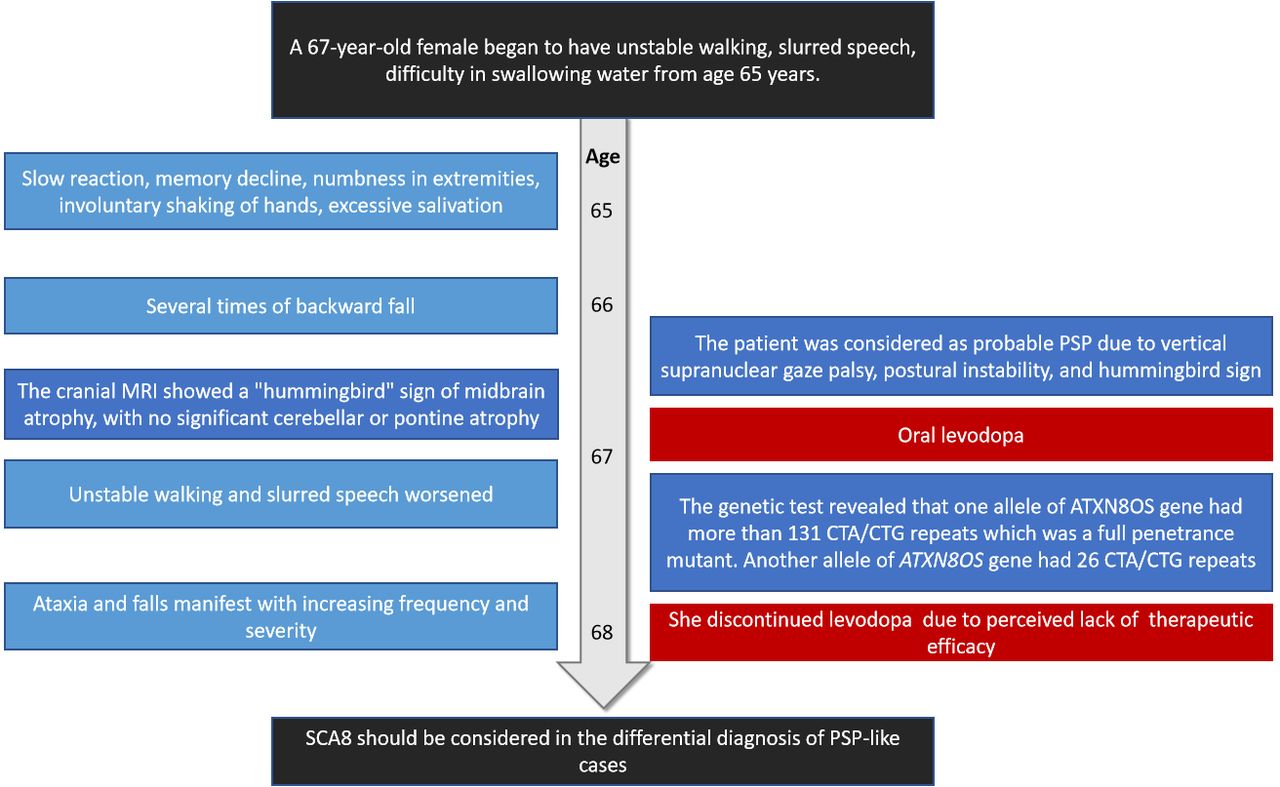
-A timeline showing the progression of patient symptoms, diagnosis, and treatment. PSP: progressive supranuclear palsy.
Diagnostic assessment
The patient was considered as probable PSP due to vertical ocular motor abnormalities, gait disturbance resulting in falls within the first two years of disease onset, and hummingbird sign. Although the patient has no positive family history, the possibility of one of the parents carrying the mutation with a late onset cannot be ruled out because the parents died at an early age (50-60 years) and the cause of the disease is unknown. Therefore, we obtained peripheral blood samples from the patient with written informed consent and extracted genomic DNA to detect SCA causing mutations in ATXN1 (SCA1), ATXN2 (SCA2), ATXN3 (SCA3), CACNA1A (SCA6), ATXN7 (SCA7), ATXN8OS (SCA8), PPP2R2B (SCA12), TBP (SCA17), ATN1 (DRPLA), NOP56 (SCA36) through polymerase chain reaction (PCR) and capillary electrophoresis. The primer is designed by Primer-Blast (primer sequences are provided in Table 1). The PCR amplification was used to amplify targeted genes. The experiment was carried out in a reaction mixture with a final volume of 10 μl PCR amplifications, which included 1μl gDNA, 7μl KOD FX Mix (Hangzhou Cred Technology Co., Ltd, hangzhou, China) and 2μl primers. The PCR program was showed in Figure 1B. The PCR products were sized by agarose gel electrophoresis and capillary electrophoresis on the Applied Biosystems 3730XL DNA Analyzer.
- Primer sequences used in PCR for this study.
The genetic test revealed that one allele of ATXN8OS gene had more than 131 CTA/CTG repeats which was a full penetrance mutant, leading to the final diagnosis of SCA 8 (Figure 1C). Another allele of ATXN8OS gene had 26 CTA/CTG repeats.
Therapeutic intervention
The patient was discharged with a prescription for oral levodopa 100mg 3 times daily.
Follow-up and outcome
The patient’s ataxia became more pronounced 2 months after discharge. Despite being prescribed levodopa and selegiline to alleviate symptoms and slow disease progression, she discontinued the medications a month later due to perceived lack of efficacy.
Discussion
According to the clinical diagnostic criteria of PSP, this patient was diagnosed with clinically probable PSP with Richardson’s syndrome (O1+P1).3 This case suggested that patients with clinical manifestations of PSP can carry repeated mutations in the ATXN8OS gene. Patients presenting with PSP carrying ATXN8OS gene mutations have been reported in two cases from Japan but have not been reported in other populations, so this study provides further evidence supporting the PSP phenotype associated with abnormal CTA/CTG repeats in ATXN8OS gene.
The SCA8 is characterized clinically mainly by onset at the age of 40-50 years, with slow progression and gradual unstable walking over 20 years. The severity of the disease is highly variable and correlates with the number of the repeat mutation. Ataxia presents in almost all the patients and dysarthria may be the earliest manifestation.1 Other symptoms include sensory impairment, sphincter dysfunction, oculomotor palsy, extra pyramidal symptoms and cognitive decline.4 Some patients carrying CTA/CTG amplification also present with multiple system atrophy type C (MSA-C) and paroxysmal kinesigenic dyskinesia.4
The neuroimaging findings of SCA8 are rarely described. Cerebral MRI reveals cerebellar vermis and hemispheric atrophy with minimal or no involvement of the brainstem, cerebral hemispheres, and basal ganglia. The MRI in this case was consistent with PSP presentation, but no significant cerebellar atrophy was observed, which needs to be confirmed by continued follow-up.
The PSP-phenotype was firstly reported to carry pathogenic mutations in ATXN8OS gene in Japanese population. ATXN8OS gene had 131 repeats (CTA10CTG121) and 93 repeats (CTA16CTG1CTA1CTG75) in patients with age at onset 76 and 67 years, respectively.5 Both of them exhibited evident parkinsonism, but neither had cerebellar ataxia nor a family history. Sporadic cases of SCA8 type are also quite common and may be mainly due to incomplete penetration. The CTA/CTG repeat sequence exhibits instability upon transmission and has a tendency to increase in maternal inheritance and decrease in paternal inheritance.6 There is significant uncertainty in the pathogenic range and incomplete penetrance of the ATXN8OS gene. The normal range of CTA/CTG repeat numbers in the ATXN8OS gene is between 15 and 50, while a repeat number exceeding 71 is considered to be pathogenic with full-penetrance.7 In 2018, SCA8 was reported for the first time in mainland China, and clinical and genetic analyses were performed on 3 Chinese SCA8 families. One of them carried 51 CTA/CTG repeat sequences in ATXN8OS gene, which is probably the shortest pathogenic allele of SCA8.8 This suggests that either genetic or environmental factors may influence the penetrance of ataxia patients carrying SCA8 amplification. Until now, the true pathological significance of this repeat expansion remains shrouded in mystery. It has been confirmed that tauopathy was present in 2 cases of SCA8 autopsy, one of which exhibited 4-repeat tauopathy resembling PSP.9 This finding reveals the complexity of SCA8 as a repeat expansion disease and highlights the necessity for further pathological research to elucidate the relationship between SCA8 and tauopathy.
In the present case, the patient had ataxia but no clear family history; although the number of repeats of the ATXN8OS gene CTA/CTG exceeded 74, the possibility that the mutation is not pathogenic cannot be completely excluded due to the incomplete penetration of the mutation; it is also possible that the combination of the mutation and the environment led to the ataxic phenotype. Whether the ATXN8OS gene CTA/CTG repeats cause PSP phenotype needs further investigation with larger samples and pathological findings. SCA8 should be considered as a potential differential diagnosis for PSP-like cases if there were other family members to suggest an autosomal dominant disease. However, there is insufficient support for the widespread implementation of genetic testing for the ATXN8OS gene among patients who meet the criteria for PSP diagnosis.
Conclusions
This study suggests a potential correlation between ATXN8OS gene abnormal CTA/CTG expansion and PSP phenotype. However, the current evidence is insufficient to support ATXN8OS gene testing for patients with PSP.
Acknowledgement
We are thankful to the patients who agreed to participate in this study. We thank Home for Researchers editorial team (www.home-forresearchers.com) for language editing service. This work was supported by the Research Foundation of Zhejiang Health (2023KY838).
- Received April 8, 2023.
- Revision received June 22, 2023.
- Accepted June 26, 2019.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.




{kind=link}
{kind=link}
{kind=link}