


Abstract
Primary hyperammonemic encephalopathy due to urea cycle disorders (UCD) typically manifests with episodic unresponsiveness and this clinical entity is not often included in the differential diagnosis of presumed non-convulsive status epilepticus (NCSE). However, this diagnostic consideration has therapeutic implications. In this report, we document the therapeutic importance of elucidating the specific cause of hyperammonemic encephalopathy that closely mimicked NCSE through 2 unique illustrative cases.
Primary hyperammonemic encephalopathy due to urea cycle disorders (UCD) typically manifests with episodic unresponsiveness1,2 and this clinical entity is not often included in the differential diagnosis of presumed non-convulsive status epilepticus (NCSE). However, this diagnostic consideration has therapeutic implications.1 In this report, we document the therapeutic importance of elucidating the specific cause of hyperammonemic encephalopathy that closely mimicked NCSE through 2 unique illustrative cases.
Case Report
Patient 1
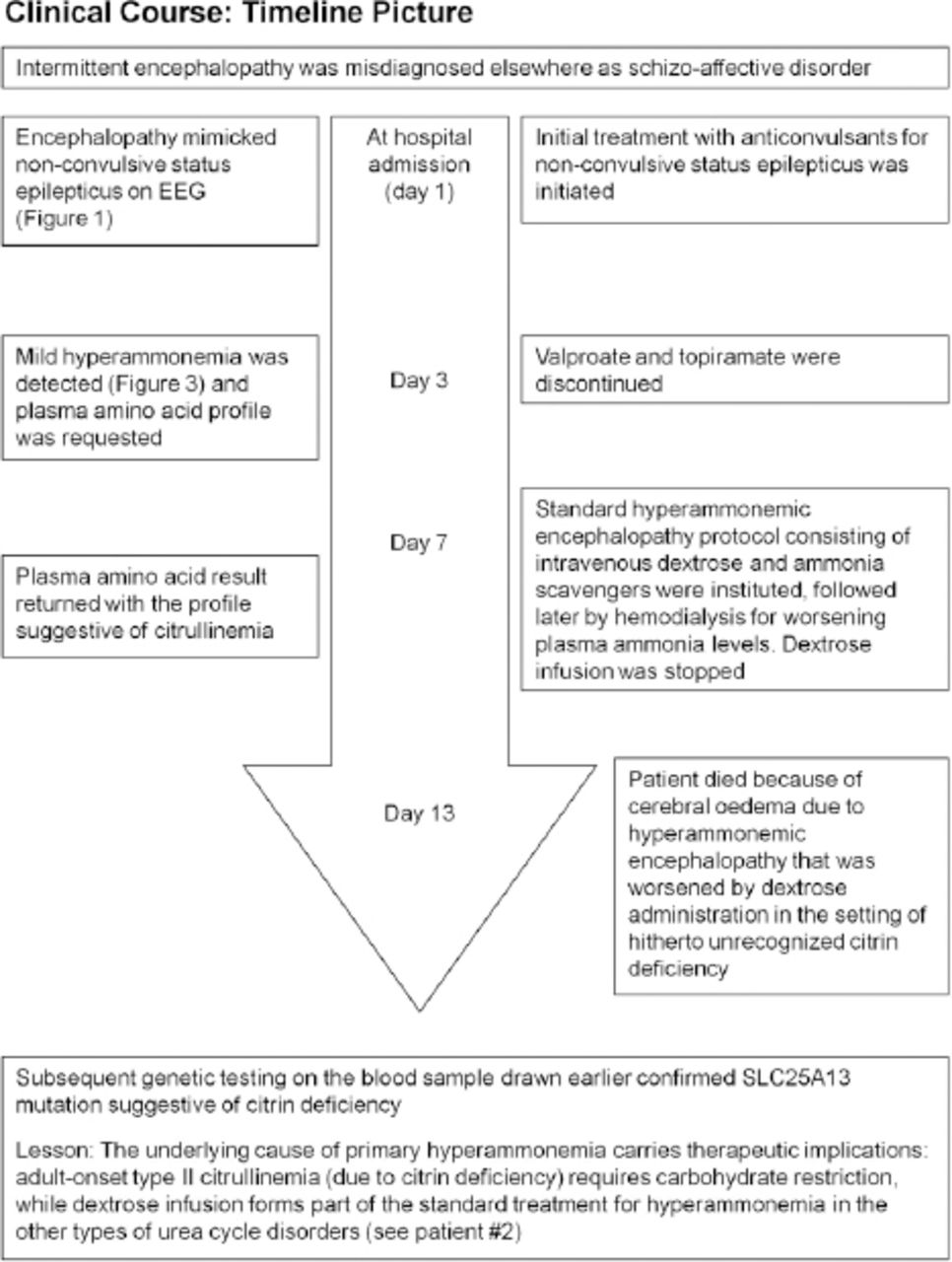
A 30-year-old woman, who was hitherto diagnosed with schizo-affective disorder and catatonia elsewhere, presented to the Sultan Qaboos University hospital, Muscat, Oman (in the year 2014) with a 5-year-history of episodic unresponsiveness (akinetic mutism) lasting for several hours to a few days with postictal amnesia; there were no apparent triggers (she did not display any particular aversion or fondness for carbohydrate or protein diet) for these episodes that occurred approximately once every 2 months. She was initially admitted under the psychiatry service [of Sultan Qaboos University Hospital (Muscat, Oman)] with such a typical episode, when an EEG was obtained. Patient information and clinical findings are shown in Figure 1.

Timeline picture of the clinical course of Patient #1(a 30-year-old woman) with hyperammonemic encephalopathy mimicking non-convulsive status epilepticus
Diagnostic assessment and therapeutic interventions
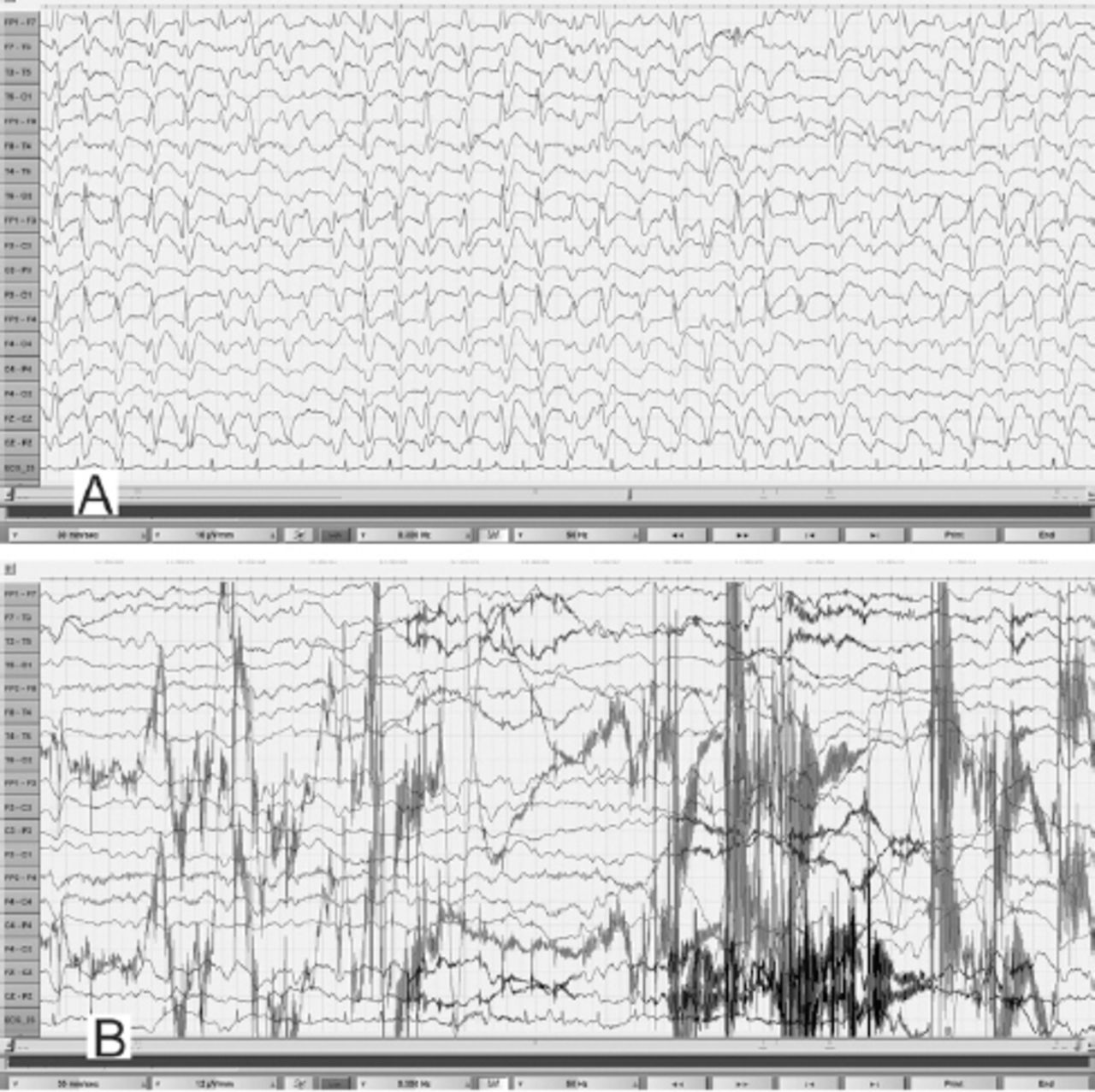
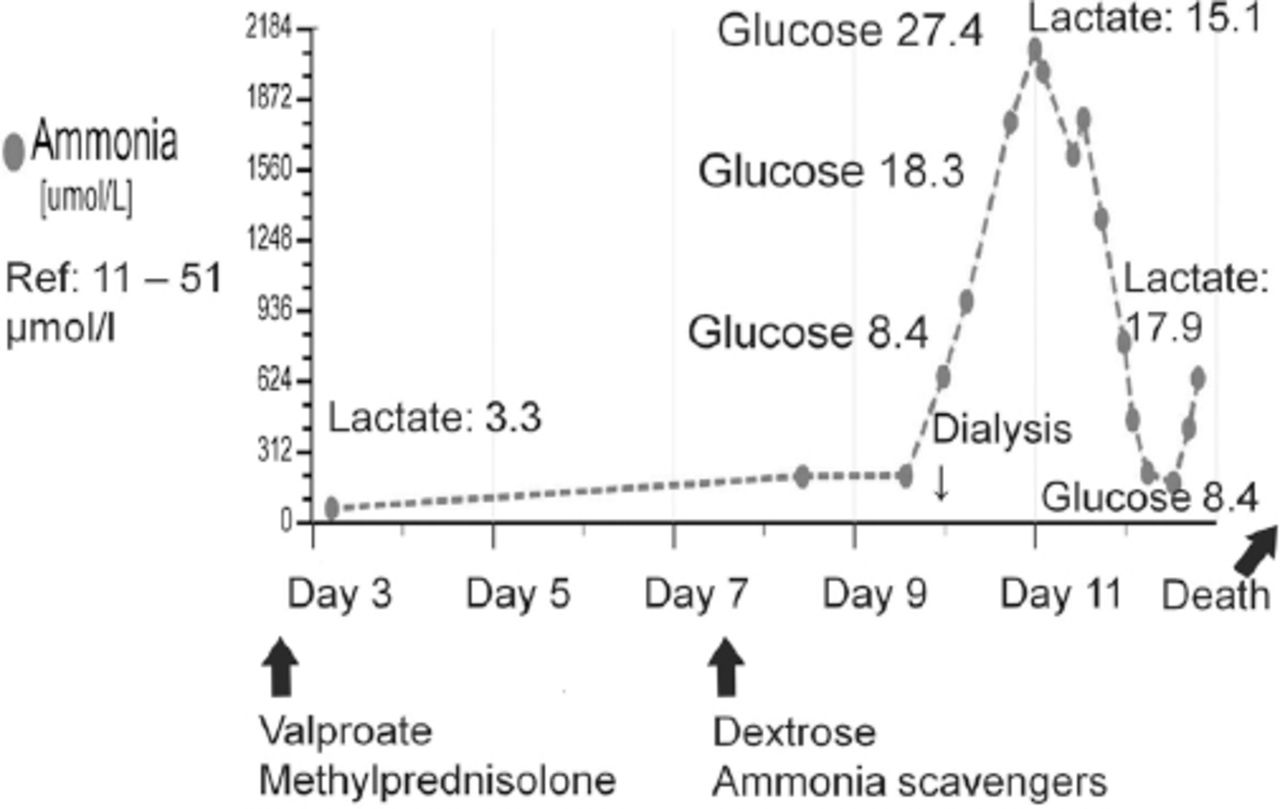
During the EEG recording (on day 3 of hospitalization), she was in coma with her eyes closed and there were continuous, generalized, 2.5 Hz sharp and slow wave discharges (Figure 2A) and she was transferred to the Neurology service. A diazepam (10 mg) trial resulted in the disappearance of the ictal sharp waves and she responded partially to painful stimuli by opening her eyes and localizing to pain (Figure 2B). There were no focal neurological deficits. In view of the continuous discharges at >2 Hz and the apparent clinical and EEG response to diazepam, a diagnosis of probable NCSE was made3 and she was placed on intravenous sodium valproate 1500 mg/day. On the subsequent day, she was again unresponsive and continuous EEG recording showed similar findings as observed in Figure 2A. However, from this moment onward, there had been no significant electro-clinical improvement with 10 mg of intravenous midazolam followed by the sequential addition of intravenous levetiracetam, phenytoin and oral topiramate in adequate doses (along with the pulse dose of methylprednisolone for presumed autoimmune encephalitis). Despite the intensive care treatment for presumed refractory NCSE, there was no appreciable clinical recovery. Her MRI brain, CSF study and autoimmune encephalitis antibody panel were unremarkable. Based on the finding of mild hyperammonemia (Figure 3) with a normal liver function test, valproate and topiramate were discontinued. While awaiting for the result of plasma amino acid profile, she was started on the standard protocol for hyperammonemic encephalopathy that consisted of 10% dextrose (with correction of extreme hyperglycemia) and intravenous sodium benzoate and sodium phenyl butyrate as ammonia scavengers. In view of the alarmingly rising hyperammonemia (Figures 1 & 3), she was also started on hemodialysis that failed to provide any definite clinical benefits. Her CT brain showed diffuse cerebral edema and repeat EEG showed nearly iso-electrical recording. At this point of time, the previously submitted plasma amino acid profile returned the following results: citrulline: 199.33 µmol/l (reference range: <55 µmol/l), arginine: 112.86 (reference range: <108.00 µmol/l), Fischer ratio=1.4 (controls: ~3.4), threonine:serine=1.8 (controls:~1.1). She was ultimately diagnosed to have adult onset type II citrullinemia (OMIM #603471) that was further confirmed genetically by the detection of homozygous deletion encompassing exon 5 of SLC25A13 gene on chromosome 7q21.3 by quantitative polymerase chain reaction.
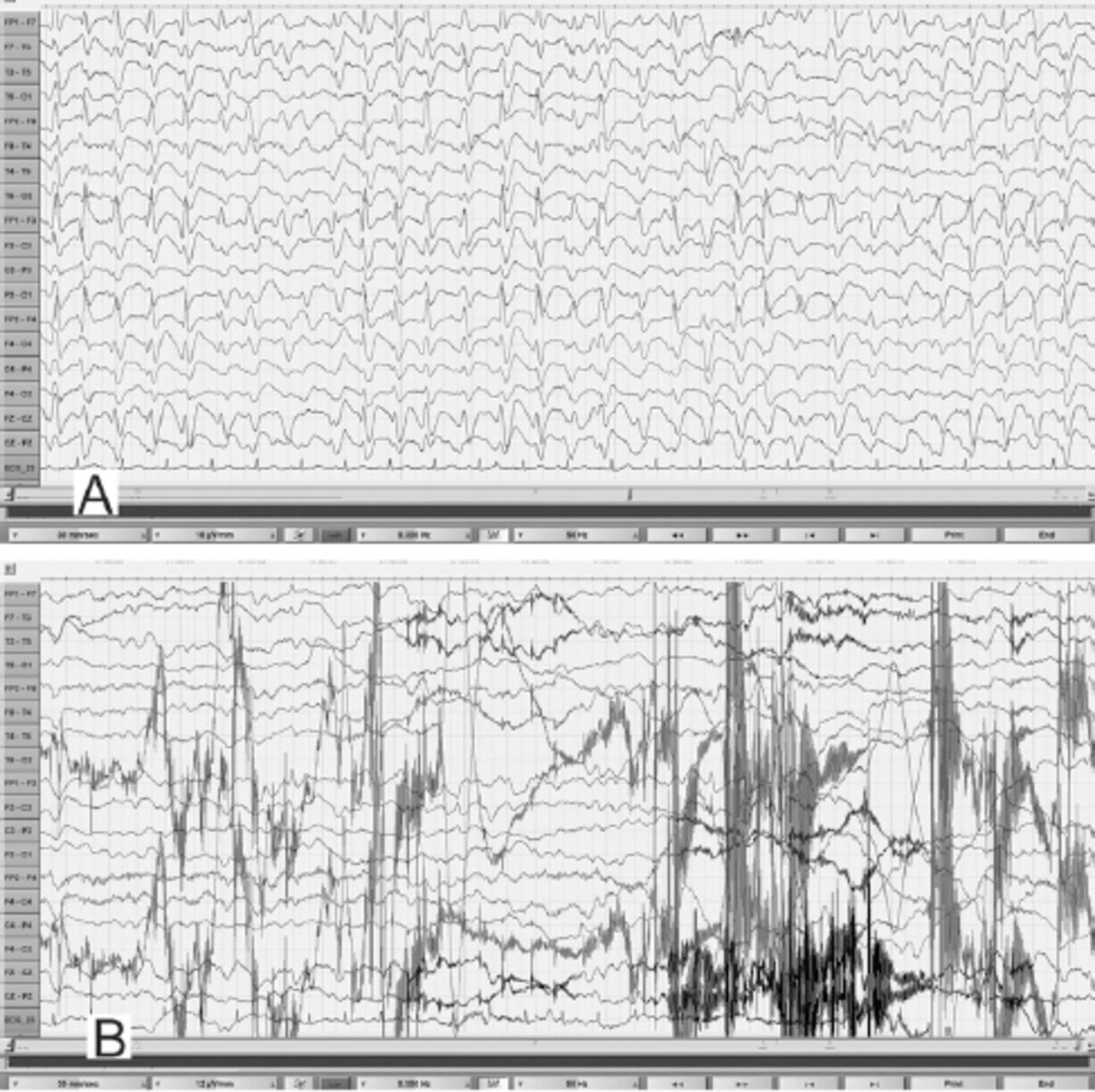
EEG obtained during an unresponsive episode (A) in a 30-year-old woman (Patient# 1) showed continuous, frontally dominant, generalized, 2.5 Hz sharp and wave discharges, with a triphasic morphology in some channels (e.g., Fz-Cz). Following intravenous administration of diazepam (B), there was complete resolution of this pattern, with the appearance of diffuse slowing in the theta range and movement artefacts (patient opened her eyes and withdrew her limbs to painful stimuli and then sat up in the bed and attempted to remove the electrodes) indicating arousal. (Low-frequency filter 0.3 Hz, high-frequency filter 50 Hz).

Marked worsening of hyperammonemia in relation to carbohydrate infusion that was provided as part of the standard treatment protocol for episodic hyperammonemic encephalopathy (mimicking non-convulsive status epilepticus) in a previously undiagnosed case (patient#1) of adult-onset citrullinemia. The plasma glucose and lactate values are in mmol/l.
Follow-up and outcome
Although dextrose infusion was discontinued, she died of worsening encephalopathy and refractory cerebral edema 15 days after admission possibly in the context of prior carbohydrate induced biochemical deterioration in citrin deficiency (Figure 3). Consent for publication has been obtained from the patient’s relative.
Patient 2
A 19-year-old man was evaluated in the Sultan Qaboos University hospital, Muscat, Oman (in the year 2014) for a 10-year-history of progressive intellectual decline, spastic paraparesis and episodic drowsiness (that was triggered by high protein diet). Patient information and clinical findings are shown in Figure 4 for the timeline picture of the clinical course of patient#2.

Timeline picture of the clinical course of Patient #2 (a 19-year-old man) with hyperammonemic encephalopathy mimicking non-convulsive status epilepticus
Diagnostic assessment
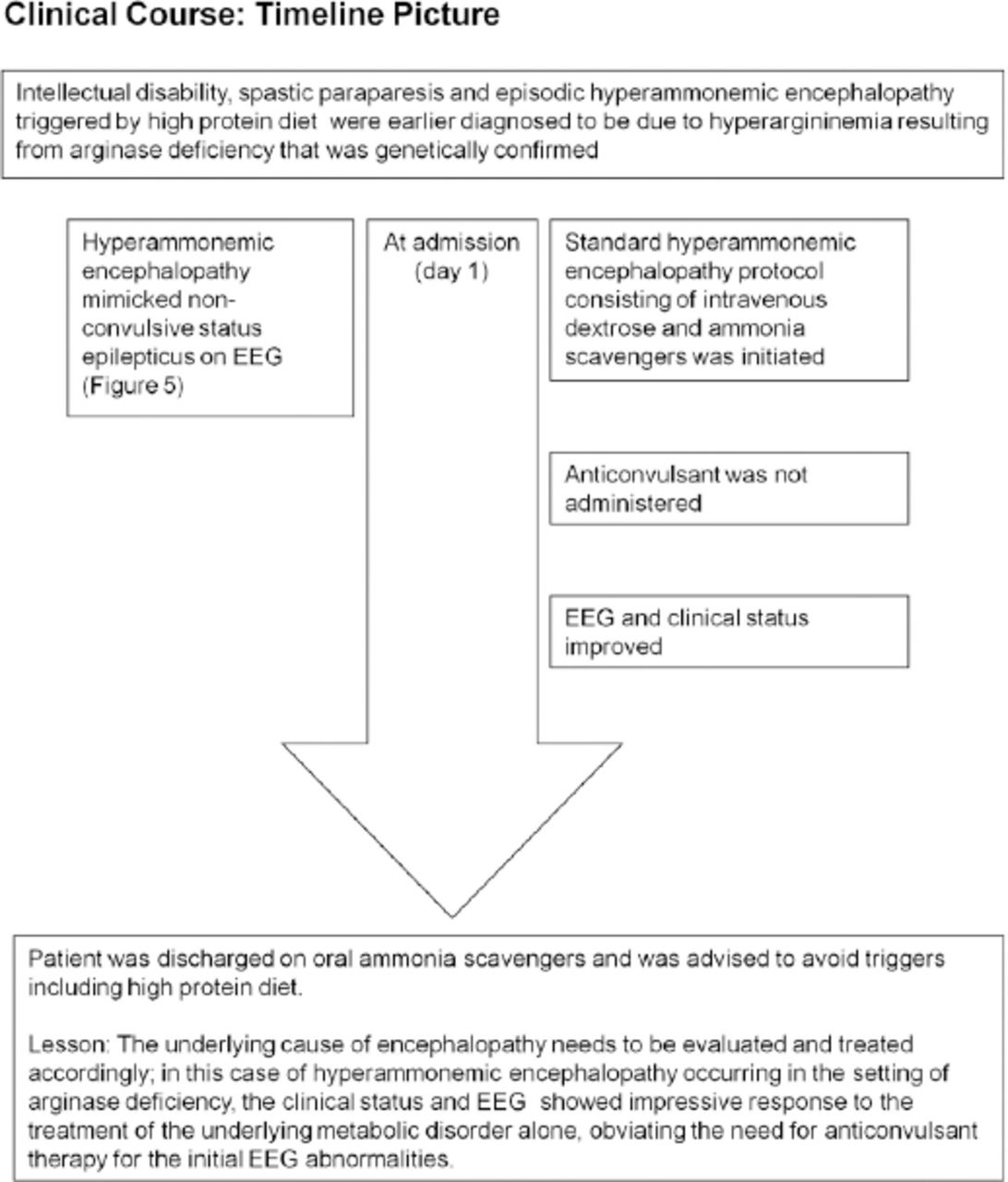
He was ultimately diagnosed to have arginase deficiency (OMIM #207800) based on the plasma amino acid profile that showed hyperargininemia (arginine: 406.85 µmol/l) and normal level of citrulline (39 µmol/l) during an episode of mild hyperammonemia (ammonia: 73.43 µmol/l; reference range: 11-51 µmol/l). Molecular genetic testing revealed a homozygous substitution c.914G>T in exon 8 of ARG1gene on chromosome 6q23.2 that resulted in the amino acid substitution p.Gly305Val. During an episode of drowsiness triggered by high protein diet, his plasma ammonia was 105 µmol/l and EEG (Figure 5A) showed identical findings as in case#1. Patient information and clinical findings are shown in Figure 4 for the timeline picture of the clinical course of patient#2.
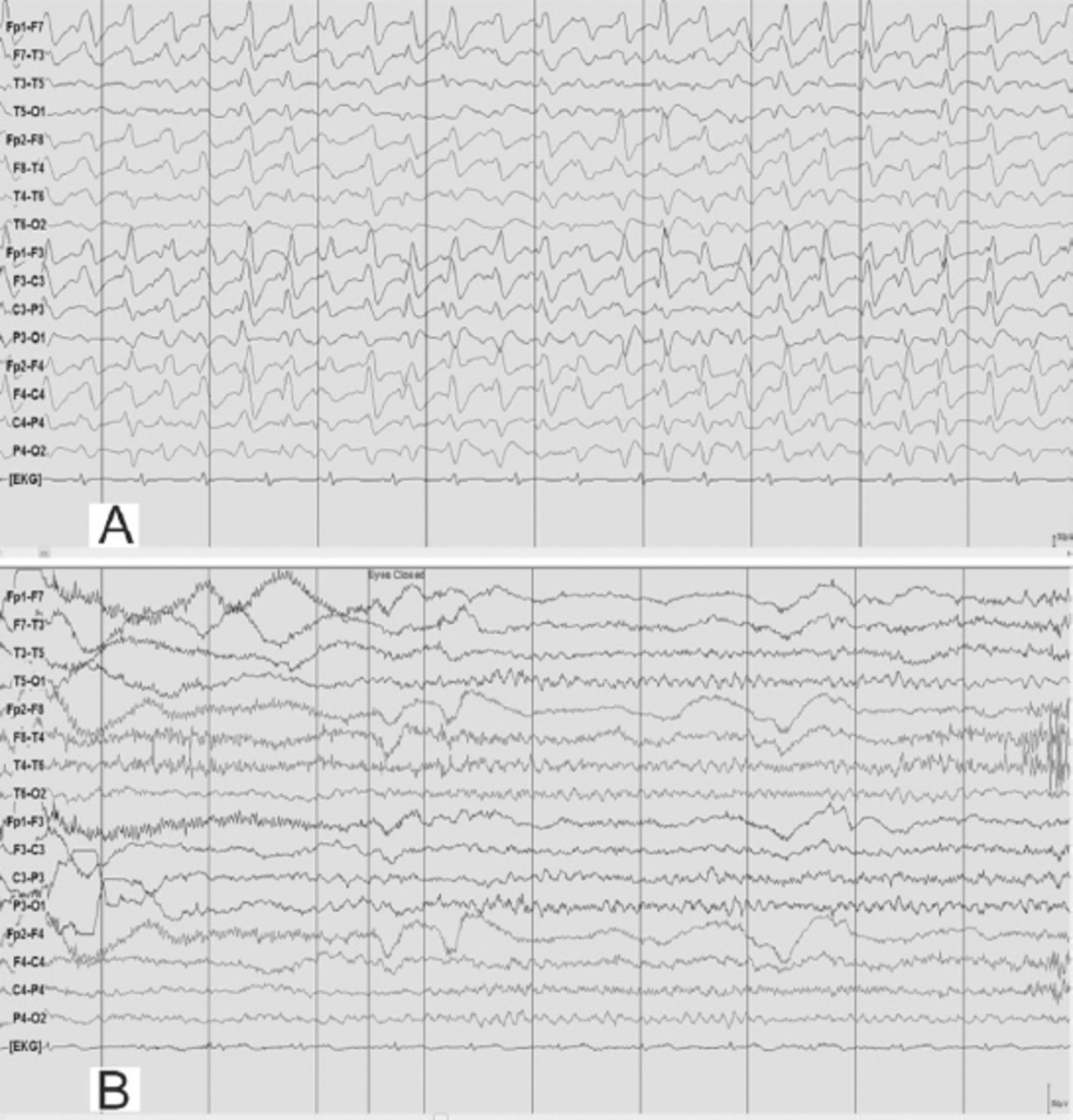
EEG obtained during an episode of hyperammonemic encephalopathy (A) with a plasma ammonia level of 105 µmol/l in a 19-year-old man (patient# 2) with a known diagnosis of arginase deficiency showed continuous, frontally dominant, generalized, 2.5 Hz sharp and wave discharges, similar to Figure 1A. Twenty hours following the standard treatment protocol for hyperammonemia that included infusion of dextrose and ammonia scavenger (B), he was conscious and alert and the corresponding EEG normalized to a posterior dominant alpha background rhythm without any ictal discharges. (Low-frequency filter 1 Hz, high-frequency filter 70 Hz).
Therapeutic intervention, follow-up and outcome
As the patient was an already diagnosed case of hyperargininemia, he was maintained on the standard hyperammonemic encephalopathy protocol consisting of intravenous dextrose and ammonia scavengers that resulted in marked clinical recovery. Pari passu, his EEG normalized to an alpha rhythm without any epileptiform, periodic or abnormal rhythmic discharges (Figure 5B). Of note, he did not receive any anticonvulsant treatment during the episode; subsequently he was continued on the oral formulation of ammonia scavengers and advised to avoid triggers such as high protein diet. The patient’s relative provided consent for publication.
Discussion
Our UCD cases (patient 1-previously undiagnosed adult-onset type II citrullinemia and patient 2- known hyperargininemia) presented with episodic hyperammonemic encephalopathy with or without obvious trigger and the electro-clinical syndrome had a close resemblance to NCSE. While the anticonvulsant therapy provided only transient electro-clinical improvement in patient 1 and was not considered in patient 2 (with the already established diagnosis of arginase deficiency), the clinical outcomes were different following the standard treatment protocol for hyperammonemia. In patient 1, in whom the rare diagnosis of adult-onset type II citrullinemia was not initially recognized, the anticonvulsant therapy was suitably modified by discontinuing sodium valproate and topiramate from the polypharmacy for presumed refractory NCSE when mild hyperammonemia was initially uncovered. However, because of her unusual electro-clinical presentation that was hitherto mistaken for catatonia and schizoaffective disorder for nearly 5 years and the latest episode being refractory to treatment with anticonvulsants, we persisted with the work-up for hyperammonemia. The timely availability of the amino acid result (not a routinely ordered biochemical test for NCSE) could have made a therapeutic difference (by the restriction of carbohydrate from the hyperammonemic encephalopathy treatment protocol in patient 1) by improving the diagnostic categorization to a specific UCD such as adult onset citrullinemia and minimizing the diagnostic and therapeutic delay.
In the context of hyperammonemic encephalopathy, pattern recognition of the amino acid abnormalities carries therapeutic implications.4,5 Thus, routine administration of dextrose should be avoided in the setting of adult onset citrullinemia, as carbohydrate loading unfavorably influences the biochemical milieu in citrin deficiency6 and this biochemical deterioration was also vividly illustrated in patient 1 (Figure 3). For the metabolic crisis of UCD mimicking NCSE, the therapeutic focus should be on improving hyperammonemia (as progressively worsening hyperammonemia has deleterious effects on brain function), instead of the overemphasis on anticonvulsant treatment alone.
To the best of our knowledge, this is the first clinical report of adult-onset type II citrullinemia from Oman. Although adult-onset citrullinemia is panethnic, there are regional differences in the reported frequency of adult-onset citrullinemia cases.7,8 For instance, a prevalence of 1:100,000-1:230,000 cases has been reported from east Asian countries such as Japan.8 At present, except for occasional case reports, population-based epidemiological studies addressing the true prevalence of adult-onset citrullinemia from other parts of the world are lacking. It is possible that this condition remains vastly under-diagnosed in many parts of the world. We hope that the present report will provide an educational impetus in raising awareness and improving the early recognition of this potentially treatable condition.
Our cases provide several learning points (take-away lessons): 1) in an otherwise unexplained encephalopathy mimicking NCSE, plasma ammonia and amino acid profile should be included in the initial laboratory panel in view of the diagnostic and therapeutic implications. Often, the genetic confirmation is not quickly available to guide the diagnostic and therapeutic pursuits at the time of metabolic crisis; 2) the standard therapeutic guideline (that excludes cases of adult-onset type II citrullinemia) suggesting dextrose infusion in primary hyperammonemic encephalopathy should be reviewed and individualized; in particular, carbohydrate restriction should be considered in the rare cases of adult-onset type II citrullinemia; 3) apart from the classical phenotype of spastic paraparesis, additional feature of hyperargininemia includes episodic hyperammonemic encephalopathy.9
We emphasize the importance of recognizing hyperammonemic encephalopathy as an imitator of NCSE, as hyperammonemia is an eminently treatable medical emergency and would prove fatal if not adequately or appropriately treated. Timely review of the amino acid profile can guide therapeutic decisions at the time of metabolic crisis.
Footnotes
Disclosure. The authors declare no conflicting interests, support or funding from any drug company.
- Received April 19, 2017.
- Accepted November 15, 2017.
- Copyright: © Neurosciences
Neurosciences is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.